Práctica XX: Aplicaciones de la bioinformática en el diseño de
Anuncio

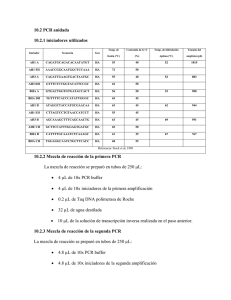
Práctica XX: Aplicaciones de la bioinformática en el diseño de estrategias experimentales. Objetivo: El alumno utilizará varias herramientas aprendidas en el curso, para el diseño de algunas aplicaciones experimentales en biología molecular. Objetivos particulares: Ø Utilizar las herramientas para la búsqueda de similitudes en secuencias para localizar la secuencia de DNA de un gen específico. Ø Conocer y aplicar los principios básicos para el diseño de iniciadores para PCR y sondas para la hibridación. Ø Emplear diversas herramientas para el diseño de iniciadores de PCR para la amplificación de secuencias individuales y un grupo de secuencias relacionadas. Ø Emplear herramientas bioinformáticas para verificar la especificidad de iniciadores. Ø Emplear herramientas de cómputo para el cálculo de mapas de corte con enzimas de restricción y el cálculo del tamaño de los fragmentos producidos. Ø Emplear herramientas bioinformáticas para visualizar propiedades de vectores de clonación y expresión. Introducción En las prácticas anteriores se ha hecho un análisis profundo de las aplicaciones más importantes que se usan comúnmente en bioinformática. No obstante en el trabajo de investigación es importante encaminar los resultados de este tipo de análisis hacia el diseño de una estrategia experimental. Hace apenas algunos años, el diseño de este tipo de estrategias se llevaba a cabo sin conocimiento previo de la secuencia de las moléculas (DNA o proteínas) en estudio. Muchas técnicas de laboratorio clásicas en bioquímica se fundamentaban exclusivamente en la actividad química de las moléculas de interés. La determinación de la secuencia o la estructura con frecuencia se llevaba a cabo hasta las últimas etapas del análisis. No obstante, con el advenimiento de las técnicas de secuenciación masiva, ahora es posible consultar primeramente la secuencia de DNA o de Proteínas de interés; incluso se dispone de un gran número de genomas secuenciados de virus, bacterias y organismos eucarióticos, por lo que ahora la búsqueda o el análisis de la secuencia tienen lugar durante las primeras etapas del diseño experimental. En torno a este tipo de estudios existen programas en el mercado que han conjuntado una gran variedad de herramientas para el análisis de secuencias para este fin, pero la gran mayoría de estas aplicaciones especializadas suelen tener costos bastante elevados. No obstante hay amplia variedad de herramientas bioinformáticas de uso libre que pueden integrarse para el diseño estrategias experimentales con fines determinados. Cabe recordar que las posibles combinaciones de estrategias experimentales en las que se pueden aplicar estos conocimientos son realmente enormes. No obstante en el análisis básico que se lleva a cabo en biología molecular para el diseño de experimentos podemos distinguir algunas técnicas importantes en las que el análisis de secuencias es fundamental. Estas aplicaciones las podemos dividir en las siguientes secciones: a) La búsqueda de las secuencias de interés (DNA o proteínas), b) el cálculo de mapas con enzimas de restricción, c) el diseño de iniciadores de PCR, d) la construcción de vectores de clonación y e) el diseño de sondas para la hibridación con microarreglos. El diseño de sondas para aplicaciones de microarreglos y el análisis de datos experimentales con esta tecnología, utiliza técnicas especiales y estos se explorarán en una práctica aparte. Entre estas aplicaciones el diseño de oligonucleótidos que puedan emplearse como iniciadores para PCR o como sondas de hibridación reviste de particular interés ya que si bien es posible seleccionar el conjunto de iniciadores sin recurrir a ningún tipo de análisis, es necesario no obstante seguir ciertas reglas para obtener iniciadores que permitan amplificar el producto con la suficiente sensibilidad y especificidad. Desde la publicación de la técnica de PCR se han formulado algunos criterios empíricos para la selección de un par de iniciadores en PCR: a) La longitud recomendable de los iniciadores debe ser de entre 17 a 24 nt. b) Deben contener un par G-C en su extremo 3’ para facilitar la iniciación de la cadena complementaria por la polimerasa. c) Cada iniciador debe poseer un contenido de G-C entre 45 y 55%. d) El iniciador no debe poseer más de cuatro pares de bases contiguos con homología a otras regiones de la cadena de DNA blanco. Los criterios anteriores pueden funcionar de manera aceptable para la amplificación de productos de entre 200 y 400 pb. Sin embargo, la amplificación de secuencias de DNA mayores es más difícil sin la aplicación estricta de otros criterios para la selección de los oligonucleótidos. Se han propuesto tres características esenciales que se deben reunir para lograr un funcionamiento óptimo como iniciadores para PCR: 1. Tanto los iniciadores como las sondas deben formar cadenas dobles estables con la cadena molde bajo las condiciones apropiadas. En general las regiones ricas en GC son más estables que las ricas en AT. Un cálculo preciso de la estabilidad del dúplex debería tomar en cuenta no solamente su composición sino la propia secuencia. 2. Los oligonucleótidos deben mostrar una especificidad muy alta por la secuencia con la que se unirán y no deben unirse más que con la región de interés. Cuando en las condiciones experimentales se produce complementariedad significativa con otras zonas de la secuencias se pueden tener resultados pobres en la amplificación del PCR y la presencia de banda indeseables que interfieren con el análisis. Es de especial interés que la complementariedad del extremo 3’ del iniciados no sea significativa con otros sitios adicionales al sitio primario de la cadena molde. 3. Los oligonucleótidos iniciadores no deben formar rizos ni dímeros durante la reacción y a que estos funcionan pobremente en varias aplicaciones e incluso puede serr particularmente indeseable cuando se aparean los extremos 3', lo cual puede causar la extensión interna del iniciador (con lo que este se elimina de la reacción) y puede contribuir a producir iniciación en falso de la replicación (false priming) en otros sitios. Para lograr estas características es conveniente a otras estrategias de diseño más complejas en las que se haga un cálculo de la estabilidad de las sondas y de su estructura secundaria con modelos termodinámicos como el del “vecino más cercano” (practica) y aplicar otras estrategias computaciones tales como Blast para asegurar su especificidad. Herramientas informáticas utilizadas. Bases de datos del NCBI (http://www.ncbi.nlm.nih.gov/). Primer3 http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi GeneFisher 2.0 http://bibiserv.techfak.uni-bielefeld.de/genefisher2/ Blast http://blast.ncbi.nlm.nih.gov BioEdit Procedimiento. I. Búsqueda de la secuencia del DNA codificante del gene 16S ribosomal en una bacteria. 1. Buscar en la base de datos del GenBank la secuencia de nucleótidos que codifica para el RNA ribosomal 16S en Escherichia coli K-12. Para esto es conveniente entrar a la página del NCBI en la sección Gene y realizar la búsqueda con la sentencia: Escherichia coli K-12 AND rrsA 2. Esta búsqueda proporcionará una lista de resultados de genes rrsA para diferentes subcepas de E. coli K-12. Para este trabajo se empleará la correspondiente a la DH10B (clave de acceso NC_010473). 3. Ingresar a los hipervínculos correspondientes y registrar las posiciones de inicio y término de esta secuencia, así como su longitud. Suponiendo que se realizará una estrategia para la amplificación de este secuencia completa, redefinir los límites de la secuencia para incluir 50 nucleótidos más al principio y 50 al final y descargar la secuencia del DNA en formato FASTA. . II. Diseño de un par de iniciadores para la amplificación de la secuencia 16S. 1. Ingresar a la herramienta primer3 para el diseño de iniciadores de uso en PCR (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) y pegar en ella la secuencia de DNA descargada en el punto anterior en la línea de captura correspondiente. 2. El programa Primer3 permite activar el diseño de iniciadores localizados cerca del extremo 5’ (left primer), en el extremo 3’ (right primer) y una sonda en el centro (internal oligo). Para estrategias de PCR simple, solo es necesario marcar las casillas correspondientes al iniciador izquierdo y el derecho. El oligonucleótido central puede emplearse para una PCR anidada o bien para el diseño de una sonda para la detección del producto por hibridación. Para este ejemplo marcar las casillas para el iniciador izquierdo y el derecho. 3. Un parámetro importante a definir es la región excluida (excluded regions). Para este ejemplo anotar 100,1400. Esto va a excluir una región de 1400 pb que comienza a partir del nucleótido 100, para la selección de iniciadores (ningún iniciador será seleccionado de esta región). Como se recordará, en el punto 1, a la secuencia del rRNA ribosomal (de aprox. 1500 nt) se le agregaron 50 pb al principio y otros 50 al final. Por lo tanto con el comando anterior, se utilizarán estas regiones para la selección de los iniciadores para la amplificación de esta secuencia más otros 50 nt de los extremos de la secuencia. De esta forma se tendrán aproximadamente 100 nt en cada extremo para la búsqueda de los iniciadores. El producto podrá tener por lo tanto una longitud entre 1400 y 1600 pb aproximadamente. Este intervalo de tamaños (1400-1600) puede especificarse en la sección “Product size ranges”. 4. Otros parámetros importantes para la selección de los iniciadores es el tamaño de los mismos (primer size), el cual se propone de 18 a 27 nt con un tamaño óptimo de 20 nt; el intervalo de Tm, el cual se propone entre 57 y 63°C con un óptimo de 60°C. Adicionalmente es conveniente especificar una diferencia máxima en el valor de Tm de 3°C. Primer3 utiliza el modelo de interacciones entre vecinos más cercanos para predecir la estabilidad de los iniciadores. No obstante no se recomienda el uso del conjunto de parámetros de Breslauer et al. (1986) que contiene bastantes errores y por lo tanto se recomienda substituirlo por el conjunto de parámetros de SantaLucia (1998) el cual es más reciente y confiable. Asimismo se recomienda la corrección de la concentración de sales propuesta por SantaLucia 1998 o bien la de Owczarzy 2004. En el programa se proponen valores de concentraciones de iones y oligonucleótidos para una PCR en condiciones estándar, sin embargo estos valores pueden cambiarse por las concentraciones empleadas en el experimento. 5. Existen además parámetros avanzados del programa los cuales no se recomienda cambiarlos durante el diseño. Es preferible modificar intervalos de selección y diferencias en Tm para lograr una selección adecuada. La opción para seleccionar una biblioteca para evitar alineamiento incorrecto de los iniciadores (mispriming library) es apropiada para la amplificación de DNA genómico eucarionte, para evitar el alineamiento de los iniciadores diseñados contra material altamente repetitivo. 6. Presionar el botón “Pick primers” para llevar a cabo la selección de los iniciadores y examinar la lista de resultados con los iniciadores calculados. Seleccionar alguna de las parejas de iniciadores de esta lista. 7. Utilizar BioEdit o GeneDoc para identificar en la secuencia la colocación de los iniciadores y estimar el tamaño del producto obtenido. 8. Realizar un análisis con Blast (empleando megablast) contra genomas bacterianos con algunos de los iniciadores seleccionados en el punto anterior para verificar que tan específicos pueden ser. Para este análisis es conveniente seleccionar solamente los organismos cuyo material genético podría estar presente en el experimento real. En este ejemplo puede seleccionar la base de datos “RefSeq_genomic” y “enterobacteria (taxid:91347)” para el organismo. La aplicación deberá mostrar un mensaje indicando que los parámetros se han ajustado para el análisis de una secuencia corta. Observe la lista de resultados obtenidos y discuta en torno a la especificidad de los iniciadores. III. Diseño de iniciadores con primer-BLAST. Primer-BLAST es una herramienta del NCBI en la cual se combinan el programa primer3 (descrito en el punto anterior) y el programa BLAST. Con esta herramienta puede verificarse la especificidad de los iniciadores durante el diseño, realizando búsquedas con BLAST contra bases de datos definidas por el usuario. El acceso a esta herramienta, puede relaizarse en la siguiente dirección: http://www.ncbi.nlm.nih.gov/tools/primer-blast/. Para el diseño debe introducirse la secuencia blanco y definir los intervalos de la secuencia de donde pueden escogerse los iniciadores (forward y reverse). Alternativamente, el usuario puede introducir secuencias predefinidas de iniciadores en cuyo caso no es necesario introducir una secuencia molde. Un paso importante es la selección de la base de datos. Para esta aplicación se pueden escoger RefSeq_mRNA, genomas ensamblados de referencia, genomas de referencia de todos los organismos, nr o incluso bases de datos personalizadas. La elección de la base de datos es importante para el análisis de especificidad y debe seleccionarse cuidadosamente tomando en cuenta si tales organismos pueden encontrarse presentes en la muestra donde se llevará a cabo la amplificación. La sección de parámetros de parámetros avanzados, muestra parámetros para configurar la búsqueda con blast (como el tamaño de palabra) y parámetros de primer3, equivalentes a los que se describieron en la sección anterior. Se deja como actividad al alumno, repetir el diseño anterior, y verificar la especificidad contra otras bacterias. IV. Diseño de un conjunto de iniciadores degenerados para amplificación de un conjunto de secuencia de DNA que codifican para el rRNA 16S de diferentes especies de bacterias. 1. Con la secuencia en el formato FASTA descargada en el punto I puede hacerse la búsqueda de secuencias similares en otras bacterias empleando las aplicaciones Blast y Megablast del NCBI. Se deja este ejercicio para el alumno. Para esta práctica se puede utilizar el conjunto de 20 secuencias de genes para rRNA 16S de bacterias en formato FASTA que puede descargarse del banco de datos del sitio WEB del curso. 2. Una vez que se ha descargado esta colección de secuencias se procederá a utilizar la herramienta GenFisher 2 (http://bibiserv.techfak.uni-bielefeld.de/genefisher2/). Presionando en “Submission” en esta página WEB se ingresa a la página de captura de la información. 3. Leer el archivo correspondiente a las secuencias de los rRNA 16S y presionar el botón “Submit”. El programa deberá mostrar un cuadro con la descripción de las 20 secuencias contenidas en el archivo. 4. El siguiente paso consiste en realizar el alineamiento de las secuencias para encontrar las regiones conservadas que permitan la selección de iniciadores para la amplificación de las 20 secuencias. Al presionar el botón “Align sequences”, se presenta un cuadro para la elección del programa para el cálculo del alineamiento. Para secuencias con un buen grado de conservación se recomiendo el empleo del programa Clustal W. Para secuencias con un bajo grado de conservación se recomienda el empleo del programa DiAlign. Para este ejemplo seleccionar el programa DiAlign. Note que las penalizaciones para la apertura y extensión de huecos pueden modificarse ¿en que casos se recomendaría modificar estos parámetros? ¿El alineamiento de las secuencias deberá ser el óptimo en este caso? Se recomienda seleccionar el coloreado basado en la similitud con la secuencia promedio. Presionar el botón para efectuar el alineamiento de las secuencias, el cual puede demorar algunos minutos en completarse. 5. Examinar el alineamiento obtenido. El coloreado por consenso puede ayudar a identificar regiones escasamente conservadas y regiones de conservación aceptable. Es conveniente que el alineamiento muestre regiones (columnas) con un grado de conservación aceptable y de longitudes apropiadas (18-25nt) para la selección de iniciadores. Si esto no se observa se recomienda repetir el alineamiento modificando las penalizaciones para la apertura y extensión de huecos o modificando el programa para el alineamiento. Para este ejemplo el alineamiento es satisfactorio por lo que se procede a presionar el botón para llevar a cabo el cálculo de la secuencia consenso. 6. Calcular el consenso con los parámetros por defecto. Realizado el cálculo la secuencia consenso se muestra en la parte superior del alineamiento. Nótese el empleo de letras para la representación de ambigüedad en la secuencia consenso. Estas letras representan la variación de bases en cada columna del alineamiento. El programa emplea dichas columnas variables para el cálculo de iniciadores degenerados en el caso de que no existan zonas de conservación total lo suficientemente grandes en el alineamiento. 7. Presionar el botón para proceder a la selección de los iniciadores. Se recomienda escoger la opción para especificar de manera manual los parámetros para el cálculo de los iniciadores. Para este caso se sugieren los siguientes valores: sin límite en el número de iniciadores, intervalo del tamaño de los iniciadores 20 a 23 pb, Tm entre 60 y 65°C. Longitud del producto entre 800 y 1000 pb. Degeneración de los iniciadores de 16 (no se recomiendan valores muy altos de degeneración, ya que esto equivale a la síntesis de una cantidad excesiva de variaciones del iniciador). No es necesario especificar que el iniciador termine en alguna base en particular, por lo que en la última sección se pueden marcar todas las bases. Presionar el botón para realizar el cálculo. Observe la lista de iniciadores obtenida (puede ser muy larga) e identifique iniciadores con secuencia definida y pares de iniciadores degenerados. Seleccione una pareja de iniciadores para ambos grupos (simples y degenerados) e identifique las regiones que amplifican con un programa tal como BioEdit o GeneDoc (deberá realizar primero el alineamiento de las secuencias). Estime además las longitudes de los productos obtenidos. 8. Realizar un análisis con Blast (empleando megablast) contra genomas bacterianos con algunos de los iniciadores seleccionados en el punto anterior para verificar que tan específicos pueden ser. Para este análisis es conveniente seleccionar solamente los organismos cuyo material genético podría estar presente en el experimento real. En este ejemplo puede seleccionar la base de datos “RefSeq_genomic” y “Bacteria (taxid:2)” para el organismo. La aplicación deberá mostrar un mensaje indicando que los parámetros se han ajustado para el análisis de una secuencia corta. Observe la lista de resultados obtenidos y discuta en torno a la posible utilización universal de los iniciadores. V. Análisis con enzimas de restricción. 1. Abrir la secuencia de rRNA 16S descargada en la sección I y emplear el programa BioEdit para editarla. El programa BioEdit ofrece varias herramientas importantes para la planeación de estrategias experimentales en biología molecular. 2. Desplegar el menú “Sequence” y búscar la opción “Nucleic Acid”. De las opciones mostradas dar clic en la opción “Restriction map”. BioEdit muestra un cuadro con varias opciones para configurar la presentación de los resultados. Algunas de las opciones más relevante permiten seleccionar que solo se muestren los resultados de enzimas que cortan un número determinado de veces, mostrar una tabla con el resumen de las frecuencias de corte, seleccionar enzimas con secuencias de reconocimiento de 4, 5 o 6 pb, presentar la traducción de la secuencias en un marco de lectura definido (solo admite el código genético universal) y la selección de enzimas específicas a partir de una lista. Estas opciones son comunes en otros programas que realizan el análisis con enzimas de restricción. 3. Para este ejemplo se recomienda activar los siguientes parámetros: Display map, Alphabetic list by name, numeric by position, sites that cut five or fewer times, 6 base cutters, enzymes with degenerate recognition. Presionar el botón “Generate map” para realizar el análisis. El programa muestra una ventana en el cual se muestra la secuencia de DNA junto con los nombres de las enzimas de restricción capaces de cortarlo y que se han colocado en el mapa en las posiciones correspondientes a los sitios de reconocimiento. Más abajo se muestra una tabla con el resumen de la frecuencia de corte para cada enzima de restricción, una lista de posiciones de reconocimiento para cada enzima y un resumen de las enzimas que no cortan el DNA. 4. Repetir el análisis anterior pero en esta ocasión seleccionar de la lista únicamente a la enzima EeaI mediante la opción “Select from list” y activar la opción “Fasta list of fragments” para generar una lista de los fragmentos de DNA que se obtendrían al cortar con esta enzima. Realizar el análisis. 5. Observe la lista de fragmentos obtenidos y el cuadro de resultados del análisis. ¿Por qué el número de fragmentos mostrados parece duplicarse? 6. Guarde el archivo de los fragmentos en formato fasta y examine las secuencias con BioEdit para estimar el tamaño de los fragmentos producidos. VI. Vectores de clonación y herramientas para la representación gráfica de secuencias. 1. Buscar en la base de datos del NCBI la secuencia de DNA con clave de acceso J01749 correspondiente al vector de clonación plasmídico pBR322. A través de la anotación disponible en el registro en formato GenBank resuma en una tabla las posiciones y la cadena en las cuales se localizan los siguientes elementos sobresalientes del plásmido: a. Gen de la resistencia a tetracilina (tet). b. Gen de la resistencia a ampicilina (beta lactamasa, bla). c. Origen de replicación. 2. Descargar la secuencia completa del plásmido en el formato FASTA. 3. Ejecutar el programa BioEdit y abrir la secuencia de DNA del plásmido descargada en el punto anterior. 4. Seleccionar el nombre de la secuencia en el editor y ejecutar la opción del menú “Sequence” > “Nucleic Acid” > “Create Plasmid from Sequence”, el cual muestra el editor gráfico para el diseño de plasmados y vectores. 5. Por defecto la secuencia se muestra en formato circular. La colocación del círculo puede modificarse dando “clic” con el ratón y arrastrando el círculo en la posición deseada. Los títulos pueden editarse dando clic sobre ellos. 6. La caja de herramientas permite adicionar elementos a la imagen y es similar a la de otros editores gráficos tales como Paint o Photo Editor. Por otra parte, la mayoría de las características del plásmido pueden manipularse con el menú “Vector”. En este menú la opción “Properties”, muestra un cuadro de diálogo mediante el cual se puede modificar el tipo de secuencia (lineal o circular), el título, características de líneas, letras y las dimensiones de la imagen, mostrar sitios de multiclonación (polylinker). También permite agregar elementos al plásmido o modificar los ya existentes (Sección “Features”, los elementos existentes aparecen en la lista desplegable”. 7. Para agregar un nuevo elemento al plásmido, tal como la región codificante de un gen se puede ejecutar el menú “Vector” > “Add feature”, lo cual abre un cuadro de diálogo que permite definir el nombre del elemento, las posiciones en donde comienza y termina, y la forma de representarlo (como una caja o una flecha), las representaciones de flecha (“Normal arrow” y “Wide arrow”) són útiles para especificar la dirección en la cual se traduce la secuencia. La dirección de esta flecha depende de la numeración colocada en las posiciones del elemento: si la posición de inicia es menor que la posición final, la flecha con dirección hacia la posición de término; si las posiciones se invierten también se invierte la dirección de la flecha. Con esta herramienta agregar los genes “tet” y “bla” de acuerdo a la características anotadas del GenBank de este plásmido y representarlos como flechas para indicar la dirección en que son traducidos dichos elementos (tome en cuenta que los elementos presentes en la cadena complementaria se traducen en dirección contraria a los presentes en la cadena directa). Si se comenten errores en el elemento agreagado, este puede modificarse desde el cuadro de diálogo “Properties” del menú “Vector”, seleccionado el elemento de la lista y presionado el botón “Modif.”. 8. El menú “Vector” > “Restriction sites” muestra un cuadro de diálogo que permite incluir en el mapa a las enzimas de restricción que cortan el plásmido. De este cuadro pueden seleccionarse las enzimas deseadas. No se recomienda mostrarlas a todas ya que su número sería excesivo. Una vez mostrados los sitios, la colocación de estos elementos puede mejorarse dando clic sobre ellos y arrastrándolos hasta la posición deseada. 9. La opción “Positional Marks” permite modificar la numeración del plásmido, en la lista “Show” se muestran las posiciones indicadas actualmente en el dibujo. Esta lista puede modificarse manualmente o automáticamente especificando el número de marcas de posición equidistantes que se desea mostrar en el mapa. 10. La imagen puede grabarse como mapa de bits (bitmap), “windows metafile” o “windows enhanced metafile”, desde el menú “File” > “Export Image”. Tambien puede copiarse en estos formatos desde el menú “Edit”. Guía para el reporte de la práctica 1. Reportar en una tabla datos relativos a la secuencia descargada: organismo de origen, posición en el genoma, clave de acceso, nombre del gen, longitud de la secuencia, composición (puede emplear BioEdit para completar esta información). 2. Reportar en una tabla las parejas de iniciadores que considere más apropiadas para la amplificación específica de la secuencia en presencia de otras enterobacterias. Incluir además la secuencia y longitud del producto obtenido así como un resumen de los resultados del análisis de especificidad con Blast. 3. Reportar en una tabla las parejas de iniciadores simples y degenerados que haya seleccionado para amplificación universal de los 20 genes 16S analizados. Incluir en una tabla los datos de la longitud de los productos obtenidos y una impresión del alineamiento en el que se identifiquen claramente los oligonucleótidos seleccionados. Incluya también un resumen del análisis de los iniciadores con Blast. ¿Qué gamma de organismos puede amplificarse con estos iniciadores? 4. Reporte el mapa de restricción en la secuencia en estudio. Incluya la tabla con la distribución de frecuencias de corte para cada enzima y la lista de las enzimas que no cortan la secuencia. Discuta en que aplicaciones puede ser importante escoger enzimas con una frecuencia elevada de corte y en que casos enzimas de baja frecuencia o que no corten a la secuencia en estudio. 5. Reporte el mapa del plásmido pBR322 indicando las características más sobresalientes que fueron anotadas en esta práctica. Preguntas. 1. Defina los términos especificidad y sensibilidad para una prueba de análisis. En el caso del diseño de iniciadores de PCR o sondas para la hibridación ¿de que factores experimentales y de diseño dependen estos parámetros? 2. ¿Qué limitantes pueden tener herramientas como Blast o Fasta para asegurar la especificidad de iniciadores de PCR o sondas de hibridación? ¿Qué tipo de análisis o herramienta computacional sugeriría para realizar un análisis más eficiente de la especificidad? 3. Un parámetro importante para lograr una buena sensibilidad y especificidad en la amplificación es evitar la formación de dímeros y rizos en los iniciadores. Las herramientas empleadas utilizan algunas reglas empíricas para evitar estos artefactos (investigue cuales son los parámetros en Primer3 y GeneFisher). ¿Qué aplicación de las que se han visto en el curso pueden aplicarse para realizar este tipo de análisis? (sugerencia: revise las aplicaciones para el cálculo de la estructura secundaria de ácidos nucleicos). 4. La estructura secundaria del blanco puede jugar un papel importante en la reacción de amplificación del PCR. Discuta en torno a como la estructura secundaría del molde podría interferir con la reacción ¿Qué herramienta podría emplearse para realizar este análisis? 5. En cuadro resuma las características más importantes de las enzimas de restricción en el que indique: la función biológica, cuantos tipos se han descrito, cuales son las que se utilizan con mayor frecuencia en el laboratorio de biología molecular, características de la secuencia de reconocimiento, tipos de cortes, ejemplos representativos. 6. Haga una representación a escala del electroferograma que podría obtenerse tras la digestión del fragmento de DNA estudiado en la práctica, con la enziam EaeI. 7. El sito WEB del NCBI incluye algunas herramientas importantes para el diseño de iniciadores. Investigue en que consiste las aplicaciones Primer-Blast y Electronic PCR en esta página. 8. El Blast del NCBI ofrece una herramienta especializada denominada VecScreen (http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html). Describa cual es la importancia que tiene para el desarrollo de técnicas experimentales. 9. Supongamos que se desea diseñar iniciadores que identifiquen de manera específica una sola secuencia entre un grupo de secuencias muy parecidas (es lo opuesto al diseño de iniciadores de la sección II). ¿Qué procedimiento seguiría para un diseño con estas características? Sugerencia, puede emplear Primer3 con restricciones para delimitar las secciones en la secuencia de interés, que pueden emplearse para seleccionar los iniciadores ¿Cómo identificaría estas regiones? 10. Describa en que consisten los iniciadores degenerados e investigue algunas características que se sugieren para la selección de los mismos. ¿Qué ventajas y desventajas pueden tener estos iniciadores en términos de su especificidad? Bibliografía. 1. Claverie J.M., Notredame C. (2003): Bioinformatics for dummies. For Dummies Series, Wiley Publishing New York. USA. pp:279-314. 2. Gibas C. Jambeck P (2001): Developing bioinformatics computer skills. O'Reilly & Associates. USA. 3. Giegerich R, M Folker y Schleiermacher Ch. (1996): GeneFisher - software support for the detection of postulated genes. Proc Int Conf Intell Syst Mol Biol. 4:68-77. 4. Roberts, R.J., Vincze, T., Posfai, J., Macelis, D. (2007) : REBASE--enzymes and genes for DNA restriction and modification. Nucl. Acids Res. 35: D269-D270. 5. Rozen S, Skaletsky H.J. (2000): Primer3 on the WWW for general users and for biologist programmers. En: Krawetz S, Misener S (eds) Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press, Totowa, NJ, pp 365-386. 6. Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. (2012): Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012 13, pp:134.