Neurotrox CR prosp 53412-00
Anuncio

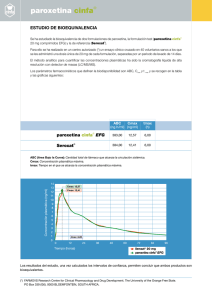
Fecha: Desarrollo Packaging 22-05-06 Motivo Emisión A P R O B A C I O N D I S E Ñ O S • T E X T O S Nº 2093/1 Producto Material Código Escala Neurotrox CR Prospecto 53412-00 100 % Correcciones solicitadas Neurotrox CR ® Paroxetina Comprimidos de liberación controlada Industria Argentina / Venta bajo receta archivada / Psicotrópico lista IV Fórmulas: Neurotrox® CR 12,5 mg: cada comprimido de liberación controlada contiene: Núcleo: paroxetina clorhidrato hemihidrato 14,22 mg (equivalente a 12,50 mg de paroxetina base). Excipientes: hidroxipropilmetilcelulosa 15,0 mg; estearato de magnesio 2,50 mg; polivinilpirrolidona K-30 10,0 mg; lactosa monohidrato USP 58,28 mg; acetoftalato de celulosa 4,50 mg; dietilftalato USP 0,31 mg; talco 0,45 mg. Cubierta: Opadry y-30-18084 4,50 mg; amarillo laca D&C N° 10 0,57 mg; amarillo laca sunset N° 6 0,03 mg; dióxido de titanio USP 1,50 mg. Neurotrox® CR 25 mg: cada comprimido de liberación controlada contiene: Núcleo: paroxetina clorhidrato hemihidrato 28,44 mg (equivalente a 25,00 mg de paroxetina base). Excipientes: hidroxipropilmetilcelulosa 18,00 mg; estearato de magnesio 2,50 mg; polivinilpirrolidona K-30 10,00 mg; lactosa monohidrato USP 71,10 mg; acetoftalato de celulosa 3,80 mg; dietilftalato USP 0,26 mg; talco 0,40 mg. Cubierta: Opadry y-3018084 3,80 mg; rojo punzó 4R 0,0092 mg; dióxido de titanio USP 1,30 mg; polivinilpirrolidona K-30 0,60 mg. Neurotrox® CR 37,5 mg: cada comprimido de liberación controlada contiene: Núcleo: paroxetina clorhidrato hemihidrato 42,68 mg (equivalente a 37,50 mg de paroxetina Base). Excipientes: hidroxipropilmetilcelulosa 23,26 mg; estearato de magnesio 5,00 mg; polivinilpirrolidona K-17 27,00 mg; lactosa monohidrato USP 193,70 mg; lauril sulfato de sodio 8,30 mg; acetoftalato de celulosa 4,50 mg; dietilftalato USP 0,30 mg; talco 0,40 mg. Cubierta: Opadry y-30-18084 4,50 mg; azul FD&C N°2 0,022 mg. Indicaciones: Neurotrox® CR, está indicada para el tratamiento de la depresión. Su eficacia se estableció en el tratamiento del episodio depresivo mayor en pacientes ambulatorios según criterios diagnósticos DSM IV. Neurotrox® CR también está indicado en trastornos de angustia con o sin agorafobia (trastorno de pánico) Neurotrox® CR, está también indicado para el tratamiento de los síntomas de los trastornos de Ansiedad Social / Fobia Social. A c c i ó n Te r a p é u t i c a : Antidepresivo. Características Farmacológicas: Acción Farmacológica: inhibidor selectivo de la recaptación de la serotonina. Paroxetina es un inhibidor de la recaptación de la serotonina (5-HT); está prácticamente desprovista de efecto directo sobre la recaptación de la noradrenalina, de la dopamina y del ácido gamaaminobutírico (GABA). Contrariamente a la mayoría de los antidepresivos tricíclicos, paroxetina no tiene prácticamente afinidad por los receptores alfa 1 adrenérgicos, histaminérgicos H1, colinérgicos (muscarínicos) D1 y D2, 5HT1 y 5HT2, α2 y β-adrenérgicos, benzodiazepínicos y opioides. Esta selectividad de la paroxetina explica la escasa incidencia de algunos efectos adversos de tipo anticolinérgicos, sedativos o hipotensión ortostática. Paroxetina exhibe las siguientes propiedades: ausencia de la alteración de la cantidad y duración del sueño, en una única toma matinal; ausencia de modificaciones de las funciones psicomotrices, ausencia de potenciación de los efectos depresores del alcohol; en voluntarios sanos, ausencia de modificaciones clínicamente significativas de la presión arterial, frecuencia cardíaca y del ECG. Farmacocinética: Neurotrox® CR controla la velocidad de liberación de la droga in vivo y tiene una cubierta entérica que demora el comienzo de la liberación de la droga hasta que el comprimido recubierto abandona el estómago. En un estudio en el cuál 23 sujetos normales recibieron dosis orales únicas de paroxetina de Liberación Controlada en concentraciones de 12,5 mg; 25 mg; 37,5 mg y 50 mg; la Cmax y el ABC 0-inf incrementaron en forma no proporcional a la dosis. Los valores medios de Cmax y AUC 0-inf con estas dosis fueron 2,0; 5,5; 9,0 y 12,5 ng/ml y 121, 261, 338, y 540 ng.hr/ml, respectivamente. El Tmax se observó entre 6 y 10 horas después de la dosificación, reflejando una reducción en la velocidad de absorción en comparación con las formulaciones de liberación inmediata. La vida media de eliminación de paroxetina fue 15 a 20 horas para todos los rangos de dosis únicas utilizadas. La biodisponibilidad de paroxetina de liberación controlada 25 mg no fue afectada por los alimentos. Durante la administración repetida de paroxetina (25 mg/día), el estado estable se alcanzó dentro de las dos semanas (es decir que fue comparable al de las formulaciones de liberación inmediata). En un estudio con dosis repetidas en 23 sujetos normales (varones y mujeres) en el cual se administró paroxetina (25 mg/día), los valores de Cmax, Cmin y AUC 024 en estado estable fueron 30 ng/ml, 20 ng/ml y 550 ng.hr/ml, respectivamente. Teniendo en cuenta los estudios realizados utilizando formulaciones de liberación inmediata, la exposición de la droga en estado estable teniendo en cuenta el AUC 0-24 fue mucho mayor que la predecible considerando los datos con dosis únicas. La acumulación en exceso se debe a que una de las enzimas encargadas de metabolizar a la paroxetina se saturan rápida y fácilmente. Los estudios de proporcionalidad de dosis en estado estable realizados en pacientes de edad avanzada y en jóvenes, con dosis de 20 a 40 mg/día de la formulación de liberación inmediata en pacientes de edad avanzada con dosis de 20 a 50 mg/día en los jóvenes, no demostraron resultados lineales en las dos poblaciones, reflejando nuevamente una vía metabólica saturable. En comparación con los valores de Cmax obtenidos después de administrar 20 mg/día, los valores de Cmax obtenidos después de 40 mg/día resultaron ser 2 a 3 veces superiores al doble. Paroxetina es extensamente metabolizada después de su administración oral. Los principales metabolitos son polares y productos conjugados de oxidación y metilación, siendo éstos rápidamente depurados. Predominan los conjugados con ácido glucurónico y sulfato. Los metabolitos principales de paroxetina han sido aislados e identificados. La potencia inhibitoria de la recaptación de serotonina de los metabolitos no supera en 1/50 a la potencia del compuesto original. El metabolismo de la paroxetina se lleva a cabo en parte a través del citocromo P450 2D6. La saturación de esta enzima, en dosis clínicas, parece ser responsable de la falta de linealidad en la cinética de la paroxetina al administrar dosis crecientes e incrementar la duración del tratamiento. El rol de esta enzima en el metabolismo de la paroxetina sugiere potenciales interacciones droga-droga. Aproximadamente el 64% de una dosis de 30 mg en solución de paroxetina oral se excreta en orina (un 2% como compuesto original y un 62% como metabolitos) durante un período de 10 días después de la administración de la droga. Aproximadamente un 36% se excreta en las heces (probablemente por vía biliar), principalmente como metabolitos, y menos del 1% como droga no metabolizada durante un período de 10 días post administración. Distribución: la distribución se realiza hacia todos los sitios del organismo, fundamentalmente hacia el sistema nervioso central, quedando sólo un 1% en el plasma. Fijación a las proteínas plasmáticas: la fijación a las proteínas plasmáticas es del 95% aproximadamente. En condiciones clínicas, los niveles de paroxetina son normalmente inferiores a 400 ng/ml. Paroxetina no modifica el enlace proteico in vitro de la fenitoína o warfarina. Pacientes con afecciones renales y hepáticas: las concentraciones plasmáticas de paroxetina incrementan en pacientes con disfunción hepática o renal. La concentración plasmática promedio en pacientes con clearance de creatinina inferior a 30 ml/min es aproximadamente cuatro veces superior que la observada en sujetos sanos. En pacientes con clearance de creatinina entre 30 y 50 ml/min y en pacientes con disfunción hepática se observa un aumento de dos veces en la concentración plasmática (ABC, Cmax). Pacientes de edad avanzada: en un estudio con dosis múltiples en ancianos, administrando dosis diarias de 20, 30 y 40 mg de la fórmula de liberación rápida, los valores de la Cmin fueron aproximadamente un 70% a 80% mayores que las respectivas Cmin en sujetos más jóvenes. En consecuencia, la dosificación inicial en ancianos se debería reducir. Posología: Trastorno Depresivo Mayor: la dosis inicial recomendada de Neurotrox® CR es 25 mg/día administrada en una única dosis diaria, por lo general, por la mañana, en ayunas o no. Neurotrox® CR en una dosis de 25 mg a 62,5 mg/día demostró se efectiva en pacientes con trastorno depresivo mayor. En aquellos pacientes que no demuestren mejoría con la dosis de 25 mg puede ser conveniente un incremento gradual a razón de 12,5 mg por día, hasta llegar a un máximo de 62,5 mg/día. Las modificaciones de las dosis deben realizarse con intervalos de una semana. La eficacia de paroxetina de liberación rápida se mantiene durante períodos de hasta 1 año con dosis de 30 mg promedio, lo que corresponde a una dosis de 37,5 mg de Neurotrox® CR, de acuerdo con las consideraciones de biodisponibilidad. Trastorno de Pánico: dosis inicial: Neurotrox® CR se debe administrar en una dosis diaria única de 12,5 mg/día. Los incrementos subsecuentes deben realizarse a razón de 12,5 mg con intervalos de al menos una semana. La dosis máxima no debe superar los 75 mg/día. Los pacientes con trastorno de pánico tratados a largo plazo con paroxetina de liberación rápida manifestaron una tasa menor de recidiva que los tratados con placebo. Los cambios en las dosis deben efectuarse con la finalidad de mantener al paciente con la dosis más baja posible. Fobia Social: dosis inicial: Neurotrox® CR se debe administrar en una dosis diaria única de 12,5 mg/día. Los incrementos subsecuentes deben realizarse a razón de 12,5 mg con intervalos de al menos una semana. La dosis máxima no debe superar los 37,5 mg/día. Dosis en pacientes ancianos o débiles y pacientes con daño renal o hepático severo: En pacientes ancianos, débiles y/o con daño renal o hepático severo la dosis inicial recomendada de Neurotrox® CR es de 12,5 mg/día. La dosis máxima no debe exceder los 50 mg/día. Modo de Administración: Los comprimidos de liberación controlada Neurotrox® CR no deben ser masticados ni triturados y se deben ingerir enteros con un poco de agua. Contraindicaciones Se contraindica el uso concomitante de Neurotrox® CR en pacientes tratados con inhibidores de la monoaminooxidasa (IMAOs) o con tioridazina. Neurotrox® CR se encuentra contraindicado en pacientes con hipersensibilidad al principio activo o a cualquier componente de la formulación. No debe ser usada en menores de 18 años con Trastorno Depresivo Mayor (DSM IV). Advertencias: Los fármacos antidepresivos pueden incrementar el pensamiento suicida y la conducta suicida en niños, adolescentes y adultos jóvenes (hasta 24 años) portadores de depresión mayor y otras patologías psiquiátricas. Cada vez que deba considerarse la utilización de paroxetina u otro fármaco antidepresivo en niños o adolescentes, deberá realizarse un balance entre la necesidad clínica, los beneficios y los potenciales riesgos. Una vez comenzado el tratamiento, deberá realizarse un control estricto de la conducta del paciente, los cambios de la misma, de su estado de humor y los indicios de ideas y/o conducta suicida. Los familiares y/o cuidadores deberán ser advertidos sobre esto y solicitarles que realicen una marcada observación y pronta comunicación acerca de los cambios de conducta y/o humor. El uso de antidepresivos con indicación aprobada por ensayos clínicos controlados con depresión mayor y otras condiciones psiquiátricas deberá establecerse en un marco terapéutico adecuado a cada paciente en particular. Esto incluye: a) que la indicación sea hecha por especialistas que puedan monitorear rigurosamente la emergencia de cualquier signo de agravamiento o aumento de la ideación suicida, como así también cambios conductuales con síntomas del tipo de agitación; b) que se tengan en cuenta los resultados de los últimos ensayos clínicos controlados; c) que se considere que el beneficio clínico debe justificar el riesgo potencial. Han sido reportados en pacientes adultos tratados con antidepresivos IRS o con otros antidepresivos con mecanismo de acción compartida tanto para el Trastorno Depresivo Mayor como para otras indicaciones (psiquiátricas y no psiquiátricas) los siguientes síntomas: ansiedad, agitación, ataques de pánico, insomnio, irritabilidad, hostilidad (agresividad), impulsividad, acatisia, hipomanía y manía. Aunque la causalidad ante la aparición de estos síntomas y el empeoramiento de la depresión y/o la aparición de impulsos suicidas no ha sido establecida existe la inquietud de que dichos síntomas puedan ser precursores de ideación suicida emergente. Los familiares y quienes cuidan a los pacientes deberían ser alertados acerca de la necesidad de seguimiento de los pacientes en relación tanto de los síntomas descriptos como de la aparición de ideación suicida y reportarlo inmediatamente a los profesionales tratantes. Dicho seguimiento debe incluir la observación diaria de los pacientes por sus familiares o quienes estén a cargo de sus cuidados. Ensayos clínicos controlados no han mostrado eficacia y no pueden sustentar el uso de Paroxetina en niños con Trastorno Depresivo Mayor (DSM IV). Paroxetina no está indicada en menores de 18 años. Si se toma la decisión de discontinuar el tratamiento la medicación debe ser reducida lo más rápidamente posible, pero teniendo en cuenta el esquema indicado para cada principio activo, dado que en algunos casos la discontinuación abrupta puede asociarse con ciertos síntomas de retirada. Pacientes que recibieron otros inhibidores de la recaptación de serotonina en combinación con IMAOS, presentaron severas reacciones (en algunos casos fatales): hipertermia, rigidez, mioclonus, inestabilidad autonómica con fluctuaciones rápidas de los signos vitales, cambios del estado mental, incluso agitación extrema, delirio y coma. Estas reacciones se reportaron en pacientes que habían discontinuando el uso de la droga e iniciado en forma inmediata el tratamiento con IMAO, por lo que se deberá respetar estrictamente un lapso de dos semanas entre la interrupción de un tratamiento con IMAOS y el inicio con paroxetina y, del mismo modo, entre el cese de administración de paroxetina y el comienzo con IMAOS. La administración de tioridazina produce una prolongación del intervalo QT, que puede asociarse con arritmias ventriculares severas, como ser arritmias del tipo "torsades de pointes" y muerte súbita. Dicho efecto aparentemente se vincula con la dosis. Un estudio in vivo sugiere que las drogas que inhiben el citocromo P450 2D6, como la paroxetina, elevan los niveles plasmáticos de tioridazina. Por lo tanto, se recomienda que la paroxetina no se utilice en combinación con tioridazina. Precauciones: En las pruebas previas a la comercialización de paroxetina, se presentaron episodios de hipomanía o manía en alrededor del 1,0% de los pacientes unipolares tratados, en comparación con el 1,1% en los casos controles tratados con drogas activas y el 0,3% de los pacientes unipolares con placebo. En un subgrupo de pacientes catalogados como bipolares, la tasa de episodios maníacos fue 2,2% con paroxetina de Liberación Controlada, y del 11,6% en los grupos combinados de control activo. Entre los 760 pacientes con trastorno depresivo mayor o trastorno de pánico tratados con paroxetina de liberación controlada en los estudios clínicos controlados, no se reportaron eventos de manía o hipomanía. Tal como ocurre con todos los fármacos efectivos para el tratamiento del trastorno depresivo mayor, Neurotrox® CR se debe utilizar con precaución en pacientes con antecedentes de manía. Durante los estudios previos a la comercialización de paroxetina de liberación rápida, se observaron convulsiones en el 0,1% de los pacientes tratados con el fármaco. Dicha tasa resulta similar a la observada con otras drogas efectivas en el tratamiento del trastorno depresivo mayor. Neurotrox® CR debe utilizarse con precaución en pacientes con antecedentes convulsivos e interrumpir su administración en pacientes que experimenten convulsiones. La posibilidad de un intento de suicidio siempre existe en el trastorno depresivo mayor y puede persistir hasta que se produzca una remisión clínica importante. La terapia farmacológica inicial debe ser estrictamente supervisada en pacientes de alto riesgo. Los estudios más recientes que sustentan las diversas indicaciones aprobadas para paroxetina de liberación rápida emplean un régimen de interrupción gradual y no abrupta del tratamiento que consiste en la paulatina reducción de la dosis diaria a razón de 10 mg utilizando intervalos semanales. Al llegar a la dosis de 20 mg diarios, los pacientes continuaron con dicha dosis durante una semana y luego se interrumpió el tratamiento. Siempre que ello sea posible, se recomienda interrumpir el uso del fármaco de manera gradual y no abrupta. En caso de que se presenten síntomas intolerables, al disminuir la dosis o al interrumpir el tratamiento, se puede considerar retomar la dosis originalmente prescripta. A continuación, el médico puede seguir disminuyendo la dosis pero en forma más gradual. Se han reportado varios casos de hiponatremia con paroxetina. La hiponatremia tendió a desaparecer al interrumpir el tratamiento con paroxetina. La mayoría de estos casos se han dado en pacientes ancianos, algunos de ellos consumían diuréticos o presentaban trastornos hidroelectrolíticos. - Embarazo: efectos teratogénicos: embarazo categoría D. Estudios preliminares indican que la paroxetina incrementa el potencial de riesgo de malformaciones congénitas. Basados en estos datos, la categoría de embarazo para paroxetina es D. Mujeres que estan siendo tratadas con paroxetina, que quedan embarazadas o estan cursando el primer trimestre de embarazo deben ser advertidas sobre los potenciales riesgos sobre el feto. La discontinuación de la terapia con paroxetina o el cambio a otro antidepresivo debe ser considerado en dichas pacientes. Uso en pacientes con enfermedades concomitantes: Con pacientes cardíacos deberán tomarse las precauciones habituales. Neurotrox® CR se debe prescribir con precaución en pacientes con glaucoma de ángulo estrecho. La evaluación de los electrocardiogramas de 682 pacientes a los que se les administró paroxetina en estudios doble ciego controlados con placebo, no demostraron que paroxetina posea relación con la aparición de anormalidades ECG significativas. De forma similar, paroxetina no ocasiona cambios importantes en el ritmo cardíaco ni en la presión sanguínea. El aumento en las concentraciones plasmáticas de paroxetina puede presentarse en pacientes con daño renal severo (depuración de creatinina <30 mL/min.) o en pacientes con daño hepático severo. En dichos pacientes debe utilizarse una dosis inicial inferior. Interacciones Medicamentosas: Triptofano: tal como ocurre con otros inhibidores de la recaptación de serotonina, el uso simultáneo de Neurotrox® CR con triptofano puede asociarse con síntomas de interacción. Se han reportado cefaleas, náuseas, sudoración y mareo. En consecuencia, no se recomienda el uso concomitante de Neurotrox® CR y triptofano. Inhibidores de la monoaminooxidasa (IMAO): el uso concomitante de inhibidores de la recaptación de serotonina con un IMAO se ha asociado con hipertermia, rigidez, mioclonías, inestabilidad autonómica con fluctuaciones rápidas de los signos vitales, alteraciones del estado mental que incluyen confusión, irritabilidad, agitación extrema, progresando a delirio y coma. El uso concomitante de Neurotrox® CR con inhibidores de la monoaminooxidasa se encuentra contraindicado, así como también dentro de los 14 días de haber discontinuado un tratamiento con un IMAO. Tampoco deberá iniciarse un tratamiento con un IMAO antes de transcurrir 14 días de interrumpido el tratamiento con Neurotrox® CR. Tioridazina: la administración de tioridazina produce una prolongación del intervalo QT, y se asocia con arritmias ventriculares severas, arritmias tipo "torsades de pointes" y muerte súbita. Dicho efecto aparentemente se vincula con la dosis. Un estudio in vivo sugiere que las drogas que inhiben el P450 2D6, tal como la paroxetina, elevan los niveles de plasma de la tioridazina. Por lo tanto, se recomienda que la paroxetina no se utilice en combinación con la tioridazina. Anticoagulantes orales (Warfarina): la administración concomitante de paroxetina puede aumentar los niveles plasmáticos de los anticoagulantes y el riesgo de sufrir hemorragias. Se recomienda precaución durante la administración concomitante con paroxetina y controlar la tasa de protrombina y el INR, y eventualmente efectuar ajustes posológicos. Pimozide: el uso de paroxetina en pacientes que estan siendo tratadas con pimozide está contraindicado. Sumatriptan: se ha informado que el uso de un inhibidor selectivo de recaptación de serotonina (ISRS) y sumatriptan puede producir debilidad, hiperreflexia e incoordinación. En consecuencia, si se considera necesario un tratamiento con sumatriptan y un ISRS se recomienda efectuar una cuidadosa vigilancia del paciente. El uso concomitante de paroxetina con otros fármacos metabolizados por el citocromo P450 2D6 aún no se ha investigado formalmente, no obstante puede ser necesario realizar un ajuste posológico y requerirse dosis más bajas de paroxetina o de los otros fármacos. Se recomienda extrema precaución cuando se administren en forma concomitante Neurotrox® CR y otros fármacos metabolizados por esta isoenzima, por ejemplo, nortriptilina, imipramina, amitriptilina, desipramina y fluoxetina, neurolépticos fenotiazínicos (como perfenazina y tioridazina), antiarrítmicos del tipo 1c, (como propafenona, flecainida y encainida) u otros inhibidores de esta enzima como la quinidina. En estado estacionario, cuando la isoenzima P450 2D6 se encuentra saturada, la depuración de paroxetina se produce a traves de otras isoenzimas P450 alternativas que no presentan saturación. Las drogas capaces de inducir o inhibir las enzimas hepáticas metabolizadoras pueden afectar los parámetros farmacocinéticos y la metabolización de la paroxetina. Durante la administración conjunta de paroxetina 30 mg/día con cimetidina oral (300 mg tres veces por día) se observó un incremento del 50% en la concentración de paroxetina. Por lo tanto, cuando estos fármacos se administren conjuntamente, la dosis inicial de 25 mg de Neurotrox® CR deberá ajustarse de acuerdo con los efectos clínicos. Se debe tener precaución al administrar antidepresivos tricíclicos (ATC) junto con Neurotrox® CR, debido a que la paroxetina puede inhibir el metabolismo de los ATC. Puede resultar necesario controlar las concentraciones plasmáticas del ATC y eventualmente reducir la dosis de los ATC si se administran junto con Neurotrox® CR. Debido a que la paroxetina presenta una alta unión a las proteínas, la administración concomitante de Neurotrox® CR con otra droga que esté muy vinculada a las proteínas puede ocasionar un incremento en las concentraciones libres de la otras drogas que también se fijan extensamente a las proteínas y ocasionar un incremento de la concentración de paroxetina libre o de la otra droga, determinando así potenciales efectos secundarios. La administración conjunta de Neurotrox® CR y litio debe realizarse con precaución. La administración conjunta de Neurotrox® CR y digoxina debe realizarse con precaución. Paroxetina aumenta la concentración plasmática de prociclidina. Si se observan efectos anticolinérgicos, se debe reducir la dosis de prociclidina. Se han informado niveles elevados de teofilina durante el tratamiento con paroxetina de liberación rápida. Si bien dicha interacción no se ha evaluado formalmente, se recomienda controlar los niveles de teofilina cuando estas drogas son administradas en forma conjunta. Tratamiento Electroconvulsivante (TEC): no existen estudios clínicos sobre el uso combinado de TEC y Neurotrox® CR. Embarazo Categoría D: ver precauciones. Trabajo de Parto y Parto: se desconoce el efecto de paroxetina durante el trabajo de parto y el parto en humanos. Madres en Etapa de Lactancia: tal como ocurre con muchos otros fármacos, la paroxetina se secreta en la leche materna, por lo tanto se desaconseja su empleo en madres que amamantan. Empleo Pediátrico: no se ha comprobado la seguridad y eficacia en la población pediátrica. Uso Geriátrico: los estudios farmacocinéticos evidenciaron una disminución de la eliminación en personas ancianas y se recomienda una dosis inicial inferior. Sin embargo, en términos generales no existieron diferencias en el perfil de eventos adversos entre pacientes ancianos y jóvenes. La efectividad fue similar en ambos casos. En un estudio controlado diseñado específicamente para pacientes ancianos con trastornos depresivos mayores, paroxetina de Liberación Controlada demostró ser segura y efectiva en dichos pacientes (> 60 de edad). Reacciones Adversas: Eventos adversos observados usualmente (incidencia del 5,0 % o mayor): Los eventos adversos más usuales asociados al uso de paroxetina de Liberación Controlada fueron: eyaculación anormal, alteraciones visuales, constipación, libido reducida, diarrea, mareos, trastornos genitales femeninos, náuseas, somnolencia, sudor, trauma, temblor, bostezos. Los eventos adversos asociados al uso de paroxetina de Liberación Controlada en pacientes ancianos fueron: eyaculación anormal, constipación, apetito disminuido, sequedad en boca, impotencia, infecciones, libido disminuida, sudoración y temblor. En el grupo de estudios realizados en trastorno de pánico, los eventos adversos correspondientes fueron: eyaculación anormal, somnolencia, impotencia, libido disminuida, temblor, sudoración y trastornos genitales femeninos (por lo general, anorgasmia o dificultad para lograr el orgasmo). Eventos adversos observados con una incidencia del 1% o mayor: Efectos adversos informados con una incidencia del 1% o mayor entre los pacientes tratados con paroxetina de liberación controlada con edades comprendidas entre 18 y 65 años con trastorno depresivo mayor que participaron en dos estudios a corto plazo controlados con placebo (12 semanas). Dichos pacientes recibieron dosis de 25 a 62,5 mg/día. Los efectos adversos producidos fueron los siguientes: Generales: cefalea, astenia, infección, dolor abdominal, dolor de espalda, trauma, dolor, reacción alérgica. Sistema cardiovascular: taquicardia, vasodilatación. Sistema digestivo: náuseas, diarrea, sequedad en boca, constipación, flatulencia, apetito disminuido, vómitos. Sistema nervioso: somnolencia, insomnio, mareo, libido disminuida, temblor, hipertonía, parestesias, agitación, confusión. Sistema respiratorio: bostezos, rinitis, aumento de la tos. Piel y anexos: sudoración, fotosensibilidad. Sentidos especiales: visión anormal, alteración del gusto. Sistema urogenital: eyaculación anormal, trastorno genital femenino, impotencia, infección del tracto urinario, trastorno menstrual, vaginitis. Eventos adversos informados con una incidencia del 5% o mayor en pacientes de 60 a 88 años con trastorno depresivo mayor, tratados con paroxetina de liberación controlada y que participaron en un estudio a corto plazo controlado con placebo (12 semanas). Estos pacientes recibieron una dosis de 12,5 a 50 mg/día. Los efectos adversos producidos fueron los siguientes: Generales: cefalea, astenia, trauma, infección. Sistema digestivo: náuseas, diarrea, sequedad en boca, constipación, flatulencia, apetito disminuido, dispepsia. Sistema nervioso: somnolencia, insomnio, mareo, libido disminuida, temblor. Piel y anexos: sudoración. Sistema urogenital: eyaculación anormal, impotencia. Efectos adversos informados con una incidencia del 1% o mayor entre pacientes de 19 a 72 años con trastorno de pánico tratados con paroxetina de liberación controlada en un estudio a corto plazo controlado con placebo (10 semanas). Este grupo de pacientes recibió una dosis de 12,5 a 75 mg/día. Los efectos adversos producidos fueron los siguientes: Generales: astenia, dolor abdominal, trauma. Sistema cardiovascular: vasodilatación. Sistema digestivo: náuseas, diarrea, sequedad en boca, constipación, apetito disminuido. Sistema nervioso: somnolencia, insomnio, nerviosismo, libido disminuida, temblor, hipertonía, ansiedad, agitación, mioclonía. Sistema respiratorio: bostezos, sinusitis. Piel y anexos: sudoración. Sentidos especiales: visión anormal, alteración del gusto. Sistema urogenital: eyaculación anormal, trastornos genitales femeninos, impotencia, frecuencia urinaria, trastornos micionales, vaginitis. Desórdenes metabólicos-nutricionales: pérdida de peso. Sistema músculo-esquelético: mialgia. Efectos adversos vinculados con la dosis: La comparación de la incidencia de efectos adversos en un estudio con dosis fija entre paroxetina y placebo en el tratamiento del trastorno depresivo mayor reveló que existe una clara vinculación entre la dosis de paroxetina de liberación rápida y algunos de los eventos más comunes. Disfunción sexual masculina y femenina con ISRS: Si bien los cambios en el deseo, el rendimiento y la satisfacción sexual por lo general ocurren como manifestaciones de un trastorno psiquiátrico, también pueden ser consecuencia del tratamiento farmacológico. Particularmente, existen evidencias que sugieren que los inhibidores selectivos de la recaptación de serotonina (ISRS) pueden inducir este tipo de experiencias sexuales negativas. Sin embargo, resulta dificultoso obtener datos confiables respecto de la incidencia y severidad de las experiencias negativas que tienen que ver con el deseo, el rendimiento y la satisfacción sexual. A continuación (Tabla 1) se presentan los porcentajes de pacientes no ancianos con trastorno depresivo mayor que reportaron síntomas de disfunción sexual en dos estudios clínicos controlados con placebo y los porcentajes correspondientes a otro estudio clínico en pacientes con trastorno de pánico. Tabla 1: Síntomas de disfunción sexual en pacientes tratados con Paroxetina de Liberación Controlada: Trastorno Depresivo Mayor Varones (n) Disminución de la libido Problemas eyaculatorios Impotencia Mujeres (n) Libido disminuida Dificultades orgásmicas Trastorno de Pánico Paroxetina de liberación controlada Placebo Paroxetina de liberación controlada Placebo 78 10 % 78 5% 1% 162 9% 194 6% 27 % 10 % 3% 1% 251 26 % 5% 134 4% 10 % 3% 133 2% <1% 282 8% 7% 2% 1% No existen estudios controlados adecuados relativos a la disfunción sexual con paroxetina. Si bien es difícil conocer el riesgo exacto de la disfunción sexual asociada con el uso de ISRS, los médicos deberían informarse con cierta regularidad de cuáles son tales posibles efectos secundarios. Cambios en el peso y en los signos vitales: La pérdida de peso en algunos pacientes puede presentarse como un resultado indeseable del tratamiento con paroxetina. Sin embargo, en los estudios clínicos controlados los pacientes tratados con Paroxetina de Liberación Controlada o con la fórmula de liberación rápida experimentaron una pérdida de peso mínima. No se observaron cambios significativos en los signos vitales (presión sanguínea sistólica y diastólica, pulso y temperatura) en los pacientes tratados con paroxetina de Liberación Controlada ni en los que recibieron la formulación de liberación rápida. Cambios electrocardiográficos: En un estudio controlado se evaluaron los registros electrocardiográficos de 682 pacientes tratados con paroxetina de liberación rápida y de 415 pacientes tratados con placebo y no se observaron cambios clínicos significativos en ningún caso. Pruebas de funcionamiento hepático: En estudios clínicos controlados con placebo, los pacientes tratados con Paroxetina de Liberación Controlada y placebo mostraron similares variaciones en los parámetros de funcionamiento hepático. No se observaron diferencias entre paroxetina y placebo con relación a las determinaciones de fosfatasa alcalina, SGOT, SGPT y bilirrubina. En un estudio clínico en pacientes ancianos con trastorno depresivo mayor, 3 de los 104 pacientes tratados con Paroxetina de Liberación Controlada y ninguno de los 109 tratados con placebo experimentaron una elevación clínicamente relevante en los niveles de transaminasas hepáticas. Dos de los pacientes tratados con paroxetina de Liberación Controlada abandonaron el estudio debido al funcionamiento hepático anormal y el tercer paciente normalizó los niveles de transaminasas al continuar el tratamiento. Asimismo, en tres estudios clínicos en pacientes con trastorno de pánico, 4 pacientes de los 444 tratados con Paroxetina de Liberación Controlada y ninguno de los 445 tratados con placebo experimentaron un incremento clínicamente relevante en los niveles de transaminasas hepáticas. El aumento de las transaminasas disminuyó de manera sustancial al suspender el tratamiento. Se desconoce el significado clínico de estos resultados. Otros eventos observados durante el desarrollo clínico de Paroxetina de Liberación Controlada: Los siguientes efectos adversos se reportaron durante el desarrollo de los estudios clínicos con paroxetina de Liberación Controlada y/o con la formulación de liberación rápida. En la etapa de evaluación previa a la comercialización de Paroxetina de Liberación Controlada se administraron dosis múltiples a 760 pacientes ambulatorios en estudios clínicos controlados doble ciego Fase III. Los efectos adversos registrados en estos estudios se clasifican por sistema corporal y se ordenan por orden decreciente de frecuencia de acuerdo con las siguientes definiciones: los efectos adversos frecuentes son los que ocurren una o más veces por lo menos en 1/100 pacientes (en esta lista sólo aparecen los que no fueron ya indicados en las pruebas clínicas controladas con placebo); los efectos adversos infrecuentes son los que se presentan entre 1/100 y 1/1.000 pacientes. Los efectos colaterales para los cuales no se indica la frecuencia se presentaron durante la etapa de evaluación pre-comercialización de paroxetina de liberación rápida en estudios de fase II y III, realizados en trastornos depresivo mayor, obsesivo-compulsivo, de pánico, de ansiedad social, de ansiedad generalizada y de estrés postraumático. Las condiciones y la duración de la exposición a la paroxetina de liberación rápida tuvieron importantes variaciones e incluyeron (en categorías comunes) estudios abiertos y doble ciego, estudios controlados y no controlados, estudios en pacientes ambulatorios y en pacientes hospitalarios y estudios de dosis fija y de titulación. A continuación se listan los efectos adversos clasificados por sistema corporal: Totalidad del cuerpo: los eventos infrecuentes fueron: reacción anafilactoide, escalofríos, síndrome gripal, malestar general; también se observó síndrome adrenérgico, edema facial, rigidez en cuello, sepsis. Sistema Cardiovascular: los eventos frecuentes fueron: hipertensión, hipotensión; los infrecuentes: angina de pecho, bradicardia, bloqueo de rama, palpitaciones, hipotensión postural, síncope; también se observaron arritmia nodal, fibrilación auricular, accidente cerebrovascular, insuficiencia cardíaca congestiva, hematoma, gasto cardíaco bajo, infarto de miocardio, palidez, flebitis, embolia pulmonar, extrasístoles supraventriculares, tromboflebitis, trombosis, cefalea vascular, extrasístoles ventriculares. Sistema Digestivo: los eventos infrecuentes fueron bruxismo, disfagia, eructos, gastroenteritis, reflujo gastroesofágico, gingivitis, glositis, hiperplasia gingival, hemorroides, hepatoesplenomegalia, sialorrea, obstrucción intestinal, melena, pancreatitis, úlcera péptica, hemorragia rectal, úlcera estomacal, dolor de muelas, estomatitis ulcerativa; además se observaron estomatitis, aftas, diarrea con sangrado, bulimia, cardioespasmo, colelitiasis, colitis, duodenitis, enteritis, esofagitis, impactación fecal, incontinencia fecal, gastritis, hemorragia gingival, hematemesis, hepatitis, ileitis, íleo, ictericia, ulceración bucal, agrandamiento de glándulas salivales, sialadenitis, estomatitis, cambios en el color de la lengua, edema de lengua. Sistema Endócrino: los eventos infrecuentes fueron hipertiroidismo, quiste de ovario, dolor testicular; también se observaron diabetes mellitus, bocio, hipotiroidismo, tiroiditis. Sistema Hemolinfático: los eventos infrecuentes fueron anemia, eosinofilia, leucocitosis, leucopenia, linfadenopatía, trombocitopenia; también se observaron anisocitosis, basofilia, aumento en el tiempo de sangrado, anemia hipocrómica, linfedema, linfocitosis, linfopenia, anemia microcítica, monocitosis, anemia normocítica, trombocitemia. Trastornos Metabólicos y Nutricionales: los eventos infrecuentes fueron bilirrubinemia, deshidratación, edema, hiperglucemia, hiperkalemia, hipokalemia, edema periférico, aumento de SGOT, aumento de SGPT, sed; también se observaron aumento de fosfatasa alcalina, aumento de BUN, aumento de creatinfosfoquinasa, elevación de gammaglobulinas, gota, hipercalcemia, hipercolesteremia, hiperfosfatemia, hipocalcemia, hipoglucemia, hiponatremia, cetosis, aumento de dehidrogenasa láctica, aumento del nitrógeno no proteico. Sistema Musculoesquelético: los efectos adversos infrecuentes fueron artritis, bursitis, miastenia, miopatía, miositis, tendinitis; también se observaron espasmos, osteoporosis, tenosinovitis, tetania. Sistema Nervioso: los eventos infrecuentes fueron amnesia, ataxia, convulsión, diplopía, distonía, inestabilidad emocional, alucinaciones, hiperestesia, hipoquinesia, incoordinación, neuralgia, neuropatía, nistagmus, parálisis, reacción paranoide, vértigo, síndrome de abstinencia; también se observaron marcha anormal, acatisia, acinesia, afasia, coreoatetosis, parestesias, delirio, disartria, discinesia, euforia, síndrome extrapiramidal, fasciculaciones, hostilidad, hiperalgesia, irritabilidad, libido aumentada, reacción maníaca, reacción maníacodepresiva, meningitis, mielitis, neuritis periférica, psicosis, depresión psicótica, disminución de reflejos, aumento de reflejos, estupor, tortícolis, trismus. Sistema Respiratorio: los eventos infrecuentes fueron asma, disnea, epistaxis, laringitis, neumonía, estridor; también se observaron disfonía, enfisema, hemoptisis, hipo, hiperventilación, fibrosis pulmonar, edema pulmonar, gripe, aumento de esputo. Piel y Anexos: los eventos infrecuentes fueron acné, alopecia, piel seca, eczema, dermatitis exfoliativa, forunculosis, prurito, seborrea, urticaria; también se observaron angioedema, equimosis, eritema multiforme, eritema nodoso, hirsutismo, exantema maculopapular, cambios en la coloración de la piel, hipertrofia cutánea, úlceras cutáneas, disminución del sudor, rash. Sentidos Especiales: los eventos infrecuentes fueron acomodación anormal, conjuntivitis, otalgia, queratoconjuntivitis, midriasis, fotofobia, hemorragia retiniana, tinnitus, defectos en el campo visual; también se observaron ambliopía, anisocoria, blefaritis, visión borrosa, cataratas, edema conjuntival, úlcera corneal, sordera, exoftalmos, glaucoma, hiperacusia, ceguera nocturna, parosmia, ptosis, pérdida del gusto. Sistema Urogenital: los eventos infrecuentes fueron albuminuria, amenorrea, agrandamiento de mamas, mastalgia, cistitis, disuria, hematuria, cálculo renal, menorragia, nocturia, protatitis, incontinencia urinaria, retención urinaria; también se observaron atrofia mamaria, alteraciones de la eyaculación, alteraciones endometriales, epididimitis, galactorrea, mama fibroquística, leucorrea, mastitis, metrorragia, nefritis, oliguria, poliuria, piuria, salpingitis, uretritis, cilindros urinarios, urgencia urinaria, urolitos, espasmo uterino, hemorragia vaginal. Diseñador Gráfico Desarrollo Packaging Control Regulatorio Revisor de texto Garantía de calidad Control Comercial Marketing Informes Post-comercialización: Los informes voluntarios de eventos adversos en pacientes tratados con paroxetina de liberación rápida que se han recibido desde su introducción al mercado y que no se indicaron más arriba y pueden no guardar relación causal con la droga son, entre otros, pancreatitis aguda, alteraciones en el funcionamiento hepático (necrosis hepática y elevación de transaminasas), síndrome de Guillain-Barré, necrólisis epidérmica tóxica, priapismo, síndrome de secreción inadecuada de hormona antidiurética, prolactinemia y galatorrea, síndrome neuroléptico maligno; síndromes extrapiramidales asociados con acatisia, bradicinesia, rigidez y rueda dentada, distonia, hipertonia, crisis oculogiras principalmente en asociación con el uso concomitante de pimozida, temblor y trismus; síndrome serotoninérgico, vinculado en ciertos casos con el uso concomitante de fármacos serotoninérgicos y con drogas que pueden alterar el metabolismo de la paroxetina (los síntomas incluyen agitación, confusión, diaforesis, alucinaciones, hiperreflexia, mioclonías, temblores y taquicardia), status epiléptico, falla renal aguda, hipertensión pulmonar, alveolitis alérgica, anafilaxis, eclampsia, neuritis óptica, porfiria, fibrilación ventricular, taquicardia ventricular (incluyendo "torsades de pointes"), trombocitopenia, anemia hemolítica y eventos relacionados con problemas hematopoyéticos (incluyendo anemia aplástica, pancitopenia, aplasia de médula ósea y agranulocitosis) y síndromes vasculíticos (tales como púrpura de Henoch-Schönlein). Se ha reportado un nivel elevado de fenitoína después de administrar durante 4 semanas paroxetina de liberación rápida concomitantemente con fenitoína y se informó hipotensión severa al agregar Paroxetina de liberación rápida al tratamiento crónico con metoprolol. Abuso de drogas y dependencia: Dependencia física y psicológica: No se ha estudiado el potencial de abuso, tolerancia o dependencia física de Neurotrox® CR en forma sistemática en animales ni en humanos. Los pacientes deben evaluarse con cuidado a fin de determinar si existen antecedentes de abuso de drogas y, en caso afirmativo, se debe efectuar un estricto seguimiento para establecer si existe un uso indebido de Neurotrox® CR (por ejemplo, desarrollo de tolerancia, aumentos en las dosis, insistencia en obtener la droga). Sobredosificación: Los eventos adversos comúnmente asociados a la sobredosis de paroxetina incluyen somnolencia, coma, náuseas, temblor, taquicardia, confusión, vómitos y mareos. Otros signos y síntomas destacables observados en casos de sobredosis con paroxetina (sola o junto con otras sustancias) incluyen midriasis, convulsiones (incluyendo status epiléptico), arritmias ventriculares (incluyendo "torsades de pointes"), hipertensión, reacciones agresivas, síncope, hipotensión, estupor, bradicardia, distonía, rabdomiolisis, síntomas de mal funcionamiento hepático (con inclusión de falla hepática, necrosis hepática, ictericia, hepatitis, y esteatosis hepática), síndrome serotoninérgico, reacciones maníacas, mioclonías, falla renal aguda y retención urinaria. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología: - Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666 / 2247 - Hospital A. Posadas: (011) 4654-6648 / 4658-7777 - Centro de Asistencia Toxicológica La Plata: (0221) 451-5555. Tratamiento: no existe un antídoto específico de la paroxetina. Estabilizar al paciente y mantener una adecuada ventilación y oxigenación; evacuación gástrica por emesis o lavado, en algunos casos puede administrarse 20 a 30 gramos de carbón activado cada 4 - 6 horas durante las primeras 24 - 48 horas posteriores a la investigación del fármaco; monitoreo cardíaco frecuente y control de signos vitales. Antagonismos. Antidotismos: No se conocen hasta el momento. Información para los pacientes: Se recomienda a los médicos tratar las siguientes cuestiones con los pacientes que inician el tratamiento con Neurotrox® CR: - Como administrar este medicamento: los comprimidos de liberación controlada de Neurotrox® CR no deben masticarse ni triturarse y deben ingerirse enteros con un poco de agua. - Interferencia con el desempeño cognitivo y motriz: todo fármaco psicoactivo puede alterar las habilidades intelectuales o motrices. Si bien en los estudios controlados Paroxetina no ha demostrado afectar el desempeño psicomotriz, se debe advertir a los pacientes que deben tener precaución al operar maquinarias peligrosas y al conducir vehículos, hasta tanto estén seguros de que el uso de Neurotrox® CR no afecta su capacidad de desarrollar tales actividades. - Cumplimiento del tratamiento: si bien es posible que los pacientes noten una mejoría durante el tratamiento con Neurotrox® CR entre la primera y la cuarta semana, se les debe recomendar continuar el tratamiento. - Medicación concomitante: los pacientes deben informar al médico si están tomando o planean tomar algún medicamento, prescripto o no, debido a que existen consecuencias posibles de interacciones con otras drogas. - Alcohol: si bien no se ha demostrado que el fármaco pueda incrementar la alteración de las facultades mentales y motoras inducidas por el alcohol, se les debe recomendar a los pacientes evitar su ingesta durante el tratamiento con Neurotrox® CR. - Embarazo: las pacientes deben informar al médico si están embarazadas o si planean estarlo durante el tratamiento. - Lactancia: las pacientes deben informar al médico si se encuentran en etapa de lactancia. - Exámenes de laboratorio: no existe ningún análisis de laboratorio que específicamente pueda ser alterado. Presentaciones: Neurotrox® CR 12,5; 25 y 37,5 mg Envases conteniendo 10, 20, 30, 40, 50, 60 y 1000 (este último para uso exclusivo hospitalario) Comprimidos de Liberación Controlada. ESTE MEDICAMENTO DEBE SER USADO EXCLUSIVAMENTE BAJO PRESCRIPCIÓN Y VIGILANCIA MÉDICA Y NO PUEDE REPETIRSE SIN NUEVA RECETA MÉDICA. Mantener éste y todos los medicamentos fuera del alcance de los niños. Conservación: Conservar en el envase original en lugar fresco y seco, protegido de la luz. No exponer a temperaturas mayores de 30°C. Esp. Med. aut. por el M.S.y A. Certificado Nº 50.702 Laboratorio Elea S.A.C.I.F.y A. Sanabria 2353. CABA. Director Técnico: Isaac J. Nisenbaum. Farmacéutico. Elaborado en Av. Gral. Juan G. Lemos 2809. Villa de Mayo. Buenos Aires. Ultima revisión: abril / 2006 53412-00 1-dil-p Control Técnico Producción Control Médico Dirección Médica Aprobación Final Dirección Técnica Fecha Proceso Películas Proc.: Firma Env.: Pantone 286 U Aclaración (Iniciales) Rec.: Obs. C A D A C O N T R O L D E B E R A R E A L I Z A R S E E N U N P L A Z O N O M AY O R D E 3 D I A S H A B I L E S