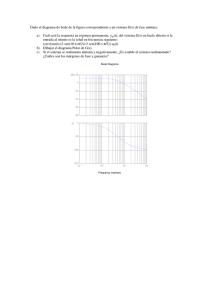
instituto tecnol´ogico y de estudios superiores de monterrey divisi
Anuncio

INSTITUTO TECNOLÓGICO Y DE ESTUDIOS SUPERIORES DE MONTERREY DIVISIÓN DE INGENIERÍA Y ARQUITECTURA APLICACIÓN DE LA TRANSFERENCIA DE IONES A TRAVÉS DE LA INTERFASE DE DOS ELECTROLITOS INMISCIBLES PARA LA IDENTIFICACION DE SALES DE AMONIO CUATERNARIO POR JOAQUÍN RODRÍGUEZ LÓPEZ PROYECTO DE INVESTIGACIÓN PRESENTADO COMO REQUISITO PARCIAL PARA OPTAR AL TÍTULO DE LICENCIADO EN CIENCIAS QUÍMICAS MONTERREY, N.L. DICIEMBRE 2005 APLICACIÓN DE LA TRANSFERENCIA DE IONES A TRAVÉS DE LA INTERFASE DE DOS ELECTROLITOS INMISCIBLES PARA LA IDENTIFICACIÓN DE SALES DE AMONIO CUATERNARIO POR JOAQUÍN RODRÍGUEZ LÓPEZ HA SIDO APROBADO DICIEMBRE 2005 COMITÉ EVALUADOR: Dr.Marcelo Videa Vargas, Asesor Dra. Leonor Blanco Jerez M.C. Marı́a Antelia de la Fuente Dávila Dr.Omar Yague Murillo ACEPTADO: Dr.Marcelo Videa Vargas Director de la Licenciatura en Ciencias Quı́micas Dr.Omar Yague Murillo Director del Departamento de Quı́mica ii RESUMEN La transferencia de iones a través de la interfase polarizable formada por dos electrolitos inmiscibles (ITIES, por sus siglas en inglés) ofrece la posibilidad de llevar a cabo el análisis cuantitativo y cualitativo de especies iónicas. El uso de técnicas electroquı́micas como la voltamperometrı́a Cı́clica (CV) ha sido exitoso para la determinación de potenciales estándar de transferencia, sin embargo sus alcances analı́ticos son limitados. Las técnicas voltamperométricas de pulso como la Voltamperometrı́a de Onda Cuadrada (SWV) proveen una aproximación diferente al problema pues permiten alcanzar menores lı́mites de detección y cuantificación, consumen significativamente menos tiempo y los voltamperogramas resultantes son más fáciles de inspeccionar y analizar. En el presente trabajo se estudió la transferencia de cationes de amonio cuaternario a través de la interfase agua/nitrobenceno mediante las técnicas de CV y SWV. Se seleccionaron los cationes de Tetrametilamonio (TMA+ ), Colina (CH+ ) y Tetrabutilamonio (TBA+ ); se encontró que el intervalo de respuesta lineal para la determinación y cuantificación de estos por SWV va de 10µM a 200µM con lı́mites de detección de 11.31µM , 7.04µM y 16.19µM y cuantificación de 37.5µM , 23.47µM y 53.95µM respectivamente. Se llevó a cabo la transferencia de iones a distintas temperaturas para evaluar el comportamiento térmico del sistema: se encontró una relación lineal entre la corriente de pico en SWV con respecto a la temperatura ası́ como una variación lineal del potencial de celda al que se observa la transferencia. iii AGRADECIMIENTOS Al Dr. Marcelo Videa por permitirme trabajar en este proyecto, por darme la oportunidad de desarrollar mis habilidades y un adquirir un carácter cientı́fico que llevaré toda la vida grabado en mi persona. Al Dr. Omar Yague por permitirnos el uso y traslado del potenciostato al laboratorio y por su participación como sinodal. A la M.C. Marı́a Antelia de la Fuente por su apoyo durante este trabajo y por su participación como sinodal. A la Dra. Luz Marı́a Martı́nez por la colaboración entre laboratorios. Al Ing. Adolfo López por su gran ayuda en los detalles de la operación del potenciostato y por su amistad como colega electroquı́mico. Al Lic. Vı́ctor Agmo por sentar las bases de este proyecto. A Don Félix Chávez por su invaluable colaboración en el soplado, construcción y reparación de la celda electroquı́mica. A la Dra. Leonor Blanco (UANL) por su participación como sinodal. A la Dra. Elsa Guajardo por la colaboración entre laboratorios. iv DEDICATORIA Con amor, a mi familia. A mis papás, Gelo y Mario por darme toda la libertad de ser, pensar y hacer lo que siento es correcto. Por darme el privilegio de ir a las mejores escuelas y de dotarme de lo necesario para ser un excelente alumno, desde enciclopedias hasta juegos de quı́mica. Por su cariño, su esfuerzo y su comprensión. Por forjar mi carácter y hacerme, en pocas palabras, quien soy. A Nayeli e Israel por recibirme en Monterrey para estudiar y por su afecto, ayuda y comprensión. Resulta inefable lo que una vida de agradecimiento implica. A Mario por estar ahı́ cuando es importante, con sus altas y sus bajas, pero ahı́. A mis sobrinas Ana Marı́a y Sofı́a, a quienes explicar este trabajo será tal vez el mayor reto. Al Dr. Marcelo Videa y a la Dra. Luz Marı́a Martı́nez, por su apoyo incondicional como persona y como alumno. Por darme la oportunidad de desarrollarme en todos los ámbitos, incluido el de cientı́fico. Por su confianza en el desarrollo de tantos proyectos y por todas las facilidades de aprendizaje, desde el trabajo en el laboratorio hasta la asistencia a congresos. Ustedes son la piedra en la que se soporta mi formación como quı́mico, son un ejemplo de vida y trabajo al cual aspiro. A la M.C. Marı́a Antelia de la Fuente, por verme crecer como quı́mico y como persona y por siempre estar ahı́, por los incentivos para ser un cientı́fico en todo el sentido de la palabra. Al Dr. Javier Rivas por su apoyo a través de la beca “Xorge Domı́nguez”, una ayuda importantı́sima en tiempos en que sin ella tal vez esta historia de investigación no se estarı́a escribiendo. A todos mis maestros. Porque cada contribución en las condiciones iniciales tiene un impacto importante en la evolución de un sistema, y cada palabra hablada, escrita y discutida fue oro. En especial, gracias por su consejo y amistad al Dr. Bernard Micheli, al Dr. Porfirio Caballero y al Dr. Mario Álvarez. v A quienes me acompañaron durante esta etapa: a Olivia, gracias por tu amor. A Fernando, Daniel, Miguel, Paulina, Yanel, Tania, Diana, Mary y mis demás compañeros de generación. Y quienes también me acompañaron y ahora me demuestran lo bueno de haber estudiado quı́mica, gracias por su guı́a, su cariño y su ayuda: Lic.Cohinta Bocanegra, Lic. Francisco Mederos, Lic. Jesús Huacuja, Lic. Gabriela López de Lara, Lic. Marı́a Durán, Lic. Guillermo Carrillo, Lic. Vı́ctor Agmo, Lic. Ramiro Rojas, Lic. Alberto Pino, Lic. Brenda Santos, Lic. Susana Chacón, Ing. Gerardo Tadeo. Con quienes compartimos la trinchera y el quehacer diario, aquellos que nos facilitan la labor: Don Pedro, Don Félix, Don Ramiro, Don Sergio, Don Lamberto y Don Carlos. vi Índice general Índice de tablas X Índice de figuras XIII 1. Introducción 1 1.1. Transferencia de Iones a través una ITIES. . . . . . . . . . . . . . . . 1 1.1.1. Fundamentos Electroquı́micos . . . . . . . . . . . . . . . . . . 2 1.1.2. Mecanismos de transferencia de iones . . . . . . . . . . . . . . 3 1.1.3. Aplicaciones de las ITIES . . . . . . . . . . . . . . . . . . . . 5 1.2. Uso de la técnica de CV para el estudio de las ITIES . . . . . . . . . 6 1.2.1. Fundamentos de CV aplicados a las ITIES . . . . . . . . . . . 8 1.2.2. Celda electroquı́mica para el estudio de las ITIES . . . . . . . 9 1.2.3. Parámetros Electroquı́micos obtenidos a partir de CV . . . . . 11 1.2.4. Parámetros Termodinámicos obtenidos a partir de CV . . . . 11 1.3. Descripción de la técnica de SWV . . . . . . . . . . . . . . . . . . . . 12 1.3.1. Fundamentos electroquı́micos de la técnica . . . . . . . . . . . 13 vii 1.3.2. Aplicaciones y ventajas de SWV . . . . . . . . . . . . . . . . . 14 1.3.3. Potencial uso de las ITIES con propósitos analı́ticos . . . . . . 15 1.4. Análisis de sales de Amonio Cuaternario . . . . . . . . . . . . . . . . 16 1.4.1. Importancia de su determinación . . . . . . . . . . . . . . . . 17 1.4.2. Técnicas analı́ticas existentes para su determinación . . . . . . 18 1.4.3. Comportamiento de las sales de Amonio en las ITIES . . . . . 19 2. Desarrollo Experimental 21 2.1. Preparativos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 2.1.1. Preparación de sal de soporte orgánica y fases . . . . . . . . . 22 2.1.2. Elaboración de electrodos de trabajo . . . . . . . . . . . . . . 23 2.1.3. Elaboración de electrodos de referencia . . . . . . . . . . . . . 24 2.1.4. Disposición de la celda electroquı́mica . . . . . . . . . . . . . . 27 2.1.5. Arreglo experimental para variación de temperaturas . . . . . 31 2.1.6. Preparación de disoluciones . . . . . . . . . . . . . . . . . . . 32 2.2. Técnicas Experimentales . . . . . . . . . . . . . . . . . . . . . . . . . 34 2.2.1. Compensación IR . . . . . . . . . . . . . . . . . . . . . . . . . 35 2.2.2. Voltamperometrı́a Cı́clica (CV) . . . . . . . . . . . . . . . . . 37 2.2.3. Voltamperometrı́a de Onda Cuadrada (SWV) . . . . . . . . . 40 3. Resultados y Discusión 42 3.1. Preparativos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . viii 42 3.1.1. Comportamiento de las Corrientes de Fondo . . . . . . . . . . 43 3.1.2. Obtención de parámetros óptimos . . . . . . . . . . . . . . . . 49 3.2. Evaluación de la transferencia por CV . . . . . . . . . . . . . . . . . 51 3.3. Evaluación de la transferencia por SWV . . . . . . . . . . . . . . . . 54 3.3.1. Transferencia de TMA+ . . . . . . . . . . . . . . . . . . . . . 55 3.3.2. Transferencia de CH+ . . . . . . . . . . . . . . . . . . . . . . 58 3.3.3. Transferencia de TBA+ . . . . . . . . . . . . . . . . . . . . . . 60 3.3.4. Análisis estadı́stico de las curvas de calibración . . . . . . . . 62 3.4. Evaluación de la calidad de las curvas en SWV y empalmes . . . . . . 70 3.4.1. Análisis de curvas en SWV . . . . . . . . . . . . . . . . . . . . 70 3.4.2. Análisis de transferencias mixtas . . . . . . . . . . . . . . . . 74 3.5. Transferencia de iones a distintas temperaturas . . . . . . . . . . . . 81 3.5.1. Comportamiento de las Corrientes de Fondo . . . . . . . . . . 83 3.5.2. Impacto de la temperatura sobre Ep . . . . . . . . . . . . . . 84 3.5.3. Impacto de la temperatura sobre ∆Ip . . . . . . . . . . . . . . 87 4. Conclusiones 92 4.1. Con respecto al desempeño analı́tico de la transferencia iónica . . . . 92 4.2. Con respecto a la evaluación de señales y empalmes . . . . . . . . . . 94 4.3. Con respecto al comportamiento térmico del sistema . . . . . . . . . 95 Bibliografı́a . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97 ix Índice de tablas 1.1. Corrientes de pico adimensionales Ψnet para SWV . . . . . . . . . . . 15 1.2. ∆φ◦i y log Pi◦ para cationes seleccionados. . . . . . . . . . . . . . . . . 19 2.1. Parámetros utilizados en Compensación IR . . . . . . . . . . . . . . . 38 2.2. Parámetros utilizados en CV . . . . . . . . . . . . . . . . . . . . . . . 40 2.3. Parámetros utilizados en SWV . . . . . . . . . . . . . . . . . . . . . . 41 3.1. Relación de corrientes de fondo en SWV a distintos P H . . . . . . . . 48 3.2. LDM y LCM calculados . . . . . . . . . . . . . . . . . . . . . . . . . 65 3.3. Análisis de [TMA+ ]= 50µM con corrección. . . . . . . . . . . . . . . 68 3.4. Análisis estadı́stico para [TMA+ ]= 50µM . . . . . . . . . . . . . . . . 69 3.5. Indicadores de la calidad de las curvas de TMA+ . . . . . . . . . . . 75 3.6. Indicadores de la calidad de las curvas de CH+ . . . . . . . . . . . . . 75 3.7. Indicadores de la calidad de las curvas de TBA+ . . . . . . . . . . . . 76 3.8. R2 para la evaluación de simulaciones de empalme. . . . . . . . . . . 80 3.9. R2 para la evaluación de la señal mixta de CH+ y TMA+ . . . . . . . 80 x Índice de figuras 1.1. Mecanismos de transferencia de carga en las ITIES . . . . . . . . . . 4 1.2. Voltamperograma del sistema [Fe(CN)6 ]−3 * ) [Fe(CN)6 ]−4 . . . . . . . 7 1.3. Transferencia de TEA+ a través de una ITIES . . . . . . . . . . . . . 9 1.4. Celda electroquı́mica de 4 electrodos. . . . . . . . . . . . . . . . . . . 10 1.5. Programación de pulsos en SWV . . . . . . . . . . . . . . . . . . . . 13 1.6. Voltamperograma de SWV . . . . . . . . . . . . . . . . . . . . . . . . 14 1.7. Geometrı́a molecular de un catión de amonio cuaternario. . . . . . . . 17 1.8. Estructura de los cationes presentados en la Tabla 1.2. . . . . . . . . 20 2.1. Estructura del BTPPATPB. . . . . . . . . . . . . . . . . . . . . . . . 23 2.2. Electrodos de Trabajo de Platino . . . . . . . . . . . . . . . . . . . . 24 2.3. Electrodos de Referencia Ag/AgCl . . . . . . . . . . . . . . . . . . . 27 2.4. LLenado de la celda de cuatro electrodos . . . . . . . . . . . . . . . . 28 2.5. Configuración de las conexiones . . . . . . . . . . . . . . . . . . . . . 30 2.6. Celda electroquı́mica montada . . . . . . . . . . . . . . . . . . . . . . 31 2.7. Celda dentro de Jaula de Faraday . . . . . . . . . . . . . . . . . . . . 32 xi 2.8. Dispositivo para mediciones a distintas temperaturas . . . . . . . . . 33 2.9. Circuito equivalente de Randles para una ITIES . . . . . . . . . . . . 36 2.10. Descomposición de las soluciones por oscilación del potencial . . . . . 39 3.1. Corrientes de fondo en CV . . . . . . . . . . . . . . . . . . . . . . . . 44 3.2. Análisis de corrientes de fondo en CV . . . . . . . . . . . . . . . . . . 45 3.3. Corrientes de fondo en SWV . . . . . . . . . . . . . . . . . . . . . . . 46 3.4. Análisis de corrientes de fondo en SWV . . . . . . . . . . . . . . . . . 47 3.5. Curva maestra para la sustracción en SWV . . . . . . . . . . . . . . . 48 3.6. Sensibilidad en CV a bajas concentraciones de analito . . . . . . . . . 51 3.7. Sensibilidad en SWV a bajas concentraciones de analito . . . . . . . . 52 3.8. Transferencia de TMA+ evaluado por CV . . . . . . . . . . . . . . . . 53 3.9. Linealización de Randles-Sevcik para de TMA+ . . . . . . . . . . . . 54 3.10. Transferencia de TBA+ 150µM evaluado por CV . . . . . . . . . . . 55 3.11. Transferencia de TMA+ en SWV sin sustracción del fondo . . . . . . 56 3.12. Transferencia de TMA+ en SWV con sustracción del fondo . . . . . . 57 3.13. Linealización de ∆Ip contra [TMA+ ] . . . . . . . . . . . . . . . . . . 58 3.14. Transferencia de CH+ en SWV con sustracción del fondo . . . . . . . 59 3.15. Linealización de ∆Ip contra [CH+ ] . . . . . . . . . . . . . . . . . . . . 60 3.16. Transferencia de TBA+ en SWV sin sustracción del fondo . . . . . . 61 3.17. Transferencia de TBA+ en SWV con sustracción del fondo . . . . . . 62 xii 3.18. Linealización de ∆Ip contra [TBA+ ] . . . . . . . . . . . . . . . . . . . 63 3.19. Gráfica de Residuales para linealización de [TMA+ ] . . . . . . . . . . 64 3.20. Gráfica de Residuales para linealización de [CH+ ] . . . . . . . . . . . 65 3.21. Gráfica de Residuales para linealización de [TBA+ ]. . . . . . . . . . . 66 3.22. Evaluación de la curva en SWV para TMA+ 100µM . . . . . . . . . 72 3.23. Evaluación de la curva en SWV para CH+ 100µM . . . . . . . . . . . 73 3.24. Evaluación de la curva en SWV para TBA+ 100µM . . . . . . . . . . 74 3.25. Simulación de transferencia mixta, E2 − E1 ≤0.1 V . . . . . . . . . . 77 3.26. Simulación de transferencia mixta, E2 − E1 ≥0.15 V . . . . . . . . . . 78 3.27. Evaluación de las curvas de transferencia mixta simulada . . . . . . . 79 3.28. Transferencia mixta de CH+ y TMA+ . . . . . . . . . . . . . . . . . 81 3.29. Evaluación de transferencia mixta de CH+ y TMA+ . . . . . . . . . . 82 3.30. Corrientes de fondo a distintas temperaturas . . . . . . . . . . . . . . 83 3.31. Transferencia de TMA+ y TBA+ a distintas temperaturas . . . . . . 84 3.32. Desplazamiento del potencial de transferencia de TMA+ . . . . . . . 85 3.33. Desplazamiento del potencial de transferencia de TBA+ . . . . . . . . 86 3.34. Variación en ∆Ip para la transferencia de TMA+ . . . . . . . . . . . 88 3.35. Variación en ∆Ip para la transferencia de TBA+ . . . . . . . . . . . . 89 xiii Abreviaciones Importantes Abreviación Significado TMA+ TBA+ CH+ BTPPA+ TPB− Ag/AgCl NB Aq ITIES CV SWV ν f P H, ESW ∆Ip ∆E ∆φ ∆tr S ∆tr G Ip Ep E1/2 F R zi , n Di Tetrametilamonio Tetrabutilamonio Colina Bis(trifenil)-fosforanilidenamonio Tetrafenilborato Electrodo de Plata / Cloruro de Plata Referente a la fase de nitrobenceno Referente a la fase acuosa Interfase entre dos electrolitos inmiscibles Voltamperometrı́a Cı́clica Voltamperometrı́a de Onda Cuadrada Velocidad de barrido en CV Frecuencia en SWV Altura del pulso en SWV Intensidad de corriente diferencial de pico en SWV Diferencia de Potencial en la celda Diferencia de Potencial de Galvani Cambio de Entropı́a de tranferencia Cambio de Energı́a Libre de tranferencia Intensidad de corriente de pico en CV Potencial de pico en SWV Potencial de media onda en CV Constante de Faraday, 96485 C Constante universal de los gases, 8.314 J/mol · K Carga del acarreador Coeficiente de difusión del ion xiv Capı́tulo 1 Introducción 1.1. Transferencia de Iones a través una ITIES. Se define una interfase como la superficie de contacto entre dos o más fases. Cuando dos fases electrolı́ticas lı́quidas inmiscibles se ponen en contacto se forma una interfase cargada entre ellas debido a la diferencia de potencial electroquı́mico de las especies acarreadoras de carga (iones). La distribución de estas especies en la región de contacto da lugar a lo que que se denomina una Interfase entre Dos Soluciones Electrolı́ticas Inmiscibles o bien Interface of Two Inmiscible Electrolytic Solutions (ITIES, por sus siglas en inglés) [1]. El estudio de las ITIES inició desde los trabajos pioneros de Nernst y Riesenfeld [2] quienes estudiaron la conducción de corriente en la interfase formada por agua y fenol. Sin embargo, el mayor impulso a la investigación de éstos sistemas se dio hasta la década de 1970, cuando Gavach [3] demostró que las ITIES podı́an ser polarizadas y presentaban la transferencia de iones de una fase a otra al aplicarse un potencial; la importancia de este descubrimiento reside en que ahora podrı́an ser investigadas de manera sistemática a través de métodos electroquı́micos y obtener información por analogı́a a sistemas en los que se presentan naturalmente o bien, aplicar sus propiedades en el desarrollo de tecnologı́a. Una caracterı́stica importante en las ITIES es que la transferencia de iones estudiada a través de métodos electroquı́micos puede modelarse matemáticamente, estableciendo parámetros que pueden ser explotados para su aplicación en técnicas analı́ticas (como en los electrodos selectivos) y para el estudio de perfiles de lipofilia de iones entre otras aplicaciones (sección 1.1.3). Dada la alta resolución propia de los métodos electroquı́micos, aunada a la selectividad y diferenciación de sustancias accesible a través de las ITIES, se abre el campo para su aplicación en problemas analı́ticos. La intención del presente proyecto es valerse de estas caracterı́sticas para llegar a la implementación de un método para identificar y cuantificar sales de amonio cuaternario. 1.1.1. Fundamentos Electroquı́micos Se ha mencionado con anterioridad que una ITIES se establece entre dos soluciones electrolı́ticas, sin embargo, la identidad de los iones que componen dichas soluciones le otorga propiedades a la interfase. En el presente trabajo se asumirá que nuestro sistema presenta una interfase idealmente polarizable. Una ITIES de este tipo se puede representar como se muestra a continuación: RE1 |A1 B1 (α)kA2 B2 (β)|RE2 (1.1) RE1 y RE2 representan electrodos de referencia en las fases α y β respectivamente y A1 B1 y A2 B2 son dos sales altamente fı́licas a la fase en la que se encuentran y fóbicas a la fase contraria; las barras dobles indican la interfase entre las soluciones inmiscibles. Cuando se ponen en contacto dos soluciones electrolı́ticas inmiscibles, existe un intercambio de iones entre ambas fases hasta llegar al equilibrio electroquı́mico, es decir, cuando el potencial electroquı́mico de una especie iónica i en ambas fases es igual µ̃αi = µ̃βi 2 (1.2) Se define al potencial electroquı́mico como la suma del potencial quı́mico (de acuerdo a la ley de Nernst) y el potencial eléctrico: + RT ln aαi + zF φα µ̃αi = µ◦,α i (1.3) Donde aαi es la actividad del ion i en la fase α y φ es el potencial de Galvani (componente del potencial eléctrico). Para describir el equilibrio del ion entre las dos fases, se igualan los potenciales electroquı́micos aαi + zi F (φα − φβ ) (1.4) β ai Si la ecuación (1.4) se divide entre el término zi F se reconoce que el término de la ∆tr G◦,α→β = µ◦,β − µ◦,α = RT ln i i i izquierda corresponde al potencial estándar de transferencia del ion i de una fase a otra, entonces: RT aβ (1.5) ln αi zi F ai Al modificar el potencial de Galvani en la ITIES, se observa un cambio de actividades ∆βα φi = ∆βα φ◦i + del ion en las fases, lo cual se puede traducir como ajuste en las concentraciones del ion i en ambas fases a fin de mantener la igualdad de la ecuación 1.5; en efecto, esto ocurre experimentalmente y existe una transferencia de iones de una fase a otra. Para el caso de las sales de soporte, los potenciales de transferencia se presentan a un valor de potencial extremo ya que son muy fı́licas a una fase y muy fóbicas a la otra. Al añadir a cualquiera de las fases un ion intermedio en cuanto a su afinidad con alguna fase respecto a las sales de soporte, se puede identificar el potencial de Galvani al cual este ion se transfiere. La conclusión anterior sobre las posibilidades que abren las ITIES para determinar la transferencia de un ion da pie a su aplicación con propósitos analı́ticos. 1.1.2. Mecanismos de transferencia de iones En las ITIES se pueden presentar en general tres mecanismos de transferencia de carga, a decir, la transferencia simple de iones, la transferencia asistida de iones y la transferencia de electrones [4],[5]. 3 La transferencia simple de iones se basa teóricamente en el tratamiento ya descrito a través de la ecuación de Nernst para las ITIES (1.5). Este mecanismo se describe como reversible y sujeto a las leyes de difusión de Fick. El estudio del transporte de iones a través de dicho mecanismo es fácilmente descrito por métodos electroquı́micos, sin embargo, existen muchos detalles teóricos que aún no han sido esclarecidos satisfactoriamente, como el cambio de propiedades en los coeficientes de difusión o las energı́as de solvatación a medida que el ion atraviesa la interfase, más aún, la descripción precisa de la estructura de la ITIES a microescala aun es tema de discusión [6]. Los otros dos tipos de transferencia escapan a los alcances del presente escrito, sin embargo, son importantes ya que proporcionan un buen fundamento para el estudio de las ITIES. Cuando en una de las fases se agrega un facilitador de intercambio (ionóforo), es decir, un agente que permite la transferencia facilitada ya sea cinética o termodinámicamente, se habla de transferencia asistida. En general, estas transferencias asistidas se dan por mecanismos de acomplejamiento ya sea en las fases o en la misma interfase [7]. La transferencia de electrones se da cuando se tienen dos pares redox separados por la ITIES y cuyo principio se fundamenta en procesos de catálisis heterogénea [1]. En todos los casos descritos en la Figura 1.1 la transferencia requiere de la aplicación de un potencial de Galvani adecuado. Figura 1.1: Mecanismos de transferencia de carga en las ITIES 4 1.1.3. Aplicaciones de las ITIES Se identifican al menos 5 áreas de oportunidad importantes en las aplicaciones de las ITIES [5]: extracción electroasistida de iones en solventes, generación de termoelectricidad, catálisis interfacial, análisis y caracterización de fármacos y aplicación en sensores amperométricos. La extracción electroasistida de iones tiene aplicación principalmente en la transferencia de iones metálicos a través de una ITIES con membrana polimérica y entre sus usos más importantes figuran la recuperación de metales en aguas residuales de la industria metalúrgica y en el estudio del transporte de iones a través de membranas biológicas. La generación de termoelectricidad se fundamenta en una analogı́a con el efecto Seebeck, que consiste en el surgimiento de una diferencia de potencial cuando dos metales a diferentes temperaturas se ponen en contacto; la idea ha sido transportada a las ITIES donde se observa la transferencia de un ion a través de la interfase debido a éste cambio de potencial, causando como respuesta una corriente. La catálisis interfacial aplicada en las ITIES se entiende fácilmente en términos del mecanismo de transferencia de carga (sección 1.1.2). Una de las aplicaciones más interesantes de esta catálisis es la conversión de energı́a solar a energı́a quı́mica, que ha sido lograda mediante la adsorción de porfirinas en la interfase y su consecuente irradiación en presencia de un par redox distribuido en las fases [5]. Sin duda, son los estudios de fármacos y la fabricación de electrodos selectivos los usos más importantes que se le han dado a las ITIES. El estudio de fármacos a través de técnicas electroquı́micas en las ITIES surge como algo natural debido a la similitud que existe entre ellas y los modelos biológicos: el caso de una interfase agua/solvente inmiscible es análogo a las membranas biológicas y a los sistemas de absorción de sustancias en los sistemas vivos. Las ITIES permiten la obtención de perfiles de lipofı́lia de medicamentos, la estimación de energı́as de transferencia y la construcción de diagramas de fase de transferencia iónica en función del pH, y por lo tanto, representan una herramienta sumamente útil para la evaluación de fármacos 5 [8],[9]. Para el presente estudio, es de mayor importancia la aplicación de las ITIES con propósitos analı́ticos. El fundamento de la aplicación de la Interfase polarizable a la quı́mica analı́tica reside en que la respuesta de las técnicas electroquı́micas de exploración depende de la concentración del analito en la fase de interés (sección 1.2.3). La determinación de concentraciones de analitos en muestras ha impulsado notablemente el desarrollo de técnicas amperométricas selectivas en las que el control de la estabilidad de las ITIES, la simplificación de los arreglos experimentales y el uso de técnicas alternas (diferentes a la Voltamperometrı́a cı́clica, sección 1.2.1) ha jugado un papel importante . La determinación amperométrica de iones a través de los mecanismos tanto de transferencia simple como de transferencia asistida, ası́ como el uso de ITIES soportadas por membranas poliméricas [10] o sistemas de difusión a microescala [11] es un campo aún en desarrollo. La construcción de un método analı́tico que aproveche las propiedades analı́ticas de las ITIES es la intención del presente estudio. 1.2. Uso de la técnica de CV para el estudio de las ITIES Cuando dos soluciones electrolı́ticas inmiscibles se ponen en contacto se forma una interfase entre ellas que resulta análoga a la formada por un electrodo metálico en una solución electrolı́tica [12], lo que permite el estudio de dichas interfases a través de técnicas electroquı́micas convencionales como la Voltamperometrı́a Cı́clica, la Polarografı́a, la Cronoamperometrı́a, la Voltamperometrı́a de corriente alterna y la Voltamperometrı́a de Redisolución de Pulso Diferencial, entre otras. El estudio de un sistema electroquı́mico generalmente empieza con la aplicación de la técnica de Voltamperometrı́a Cı́clica, ya que es una de las técnicas más simples y que ofrecen mayor información en electroquı́mica. 6 La Voltamperometrı́a Cı́clica parte del monitoreo de una intensidad de corriente en función de la variación en el tiempo de un potencial. En un electrodo metálico convencional y para un proceso controlado por difusión donde se reduce una especie oxidada, se tiene que al aproximarse al potencial de reducción se observa un aumento en la corriente registrada. A medida que la especie oxidada se reduce y su concentración disminuye notablemente en la vecindad del electrodo, la reacción electroquı́mica se ve gobernada por la difusión de la especie al electrodo y entonces la corriente registrada vuelve a caer. Esto produce una gráfica de Potencial (E) contra Intensidad de corriente (I) como la mostrada en la Figura 1.2. Figura 1.2: Voltamperograma del sistema [Fe(CN)6 ]−3 * ) [Fe(CN)6 ]−4 En la figura 1.2, Ip es la intensidad de corriente en uno de los picos del voltamperograma, Ep es el potencial de pico, y E1/2 es conocido como el potencial de media onda. E1/2 se puede calcular como el potencial intermedio de la distancia entre picos ∆Ep . La corriente de pico depende de variables como la velocidad de barrido ν, el número de electrones transferidos n, el área del electrodo A, el coeficiente de difusión 7 de la especie Di y su concentración en la solución C ∗ de acuerdo a la relación de Randles-Sevcik: Ip = (2.65E05)n3/2 AD1/2 C ∗ ν 1/2 (1.6) donde Ip está en Amperes, A en cm2 , D en cm2 s−1 , C ∗ en mol/L y ν en V/s y la temperatura es 25o C. Resulta interesante notar que la técnica es cuantitativa y que una medición de un sistema reversible deberı́a dar como resultado una gráfica lineal al comparar la intensidad de corriente contra la raı́z cuadrada de la velocidad de barrido o contra la concentración. Para un sistema reversible además de cumplirse lo anterior, también se debe hallar que la distancia entre los picos ∆Ep debe ser igual a 59 mV/n y que la posición de cada uno de ellos sea independiente de la velocidad de barrido [13]. 1.2.1. Fundamentos de CV aplicados a las ITIES Los procesos electroquı́micos no se limitan solamente a la descripción de la transferencia de electrones (en reacciones Redox), sino que se aplica a todo fenómeno que involucra una transferencia de carga. Si se establece una diferencia de potencial en una interfase en equilibrio, la ecuación 1.5 indica que habrá un cambio en las actividades de una especie activa (en este caso un ion) y por lo tanto un flujo de carga a través de la interfase. El flujo de carga es interpretado en CV como una intensidad de corriente. Al variar la diferencia de potencial entre ambas fases se observa entonces una variación en la intensidad de corriente, dando como resultado un voltamperograma. Dado que se considera que la transferencia de iones es esencialmente un fenómeno reversible, las mismas condiciones descritas en el apartado anterior son aplicables [14]. En la figura 1.3 se muestra un ejemplo de un Voltamperograma tı́pico de la transferencia de iones a través de una ITIES. En la Figura 1.3 se aprecia que existe un rango práctico para observar la transferencia iónica, lo que se conoce como ventana de potencial. Dado que en la fase 8 Figura 1.3: Transferencia del catión Tetraetilamonio en la Interfase agua/1,2dicloroetano utilizando como sal de soporte orgánico BTPPATPBCl 5mM y como soporte acuoso LiCl 0.1M. Editada de referencia [10]. acuosa como en la fase orgánica se deben usar sales de soporte, el área de estudio de transferencia de un ion está gobernada por los extremos en los que los iones de dichas sales de soporte empiezan a ser transferidos de una fase a otra. Generalmente se eligen sales muy hidrofı́licas para la fase acuosa y muy lipofı́licas para la fase orgánica, de manera que la ventana de potencial se expanda, aunque en general ésta es menor a 0.7 V en promedio [13]. 1.2.2. Celda electroquı́mica para el estudio de las ITIES El diseño de la celda electroquı́mica para llevar a cabo mediciones de las ITIES en la disposición de cuatro electrodos permite establecer la diferencia de potencial en la interfase y controlar dicho potencial a través de dos electrodos de referencia. El esquema de este arreglo se presenta en la Figura 1.4. Por simplicidad se ha considerado que las fases inmiscibles son un sistema acuoso y un sistema orgánico. La caja del electrómetro generalmente está configurada para un arreglo de tres electrodos, por lo que se debe establecer una referencia a un cuarto electrodo para dar pauta a que el potencial medido corresponda a la diferencia de potencial entre 9 Figura 1.4: Celda electroquı́mica de 4 electrodos. ambas fases. Para estos fines se adopta la convención de trabajar, para la fase orgánica con el electrodo de referencia RE para la referencia y el electrodo auxiliar CE como electrodo de trabajo, y para la fase acuosa con el electrodo de trabajo W E como electrodo de trabajo y el electrodo sensible SE como electrodo de referencia. La respuesta esperada en el potenciostato es la diferencia de potencial entre las fases: ∆Eow = ∆ESE − ∆ERE = ∆Ew − ∆Eo (1.7) Los subı́ndices w y o indican la fase acuosa y orgánica respectivamente. Es importante recalcar que la resolución de la respuesta dependerá de algunas caracterı́sticas de la celda, por ejemplo, para cumplir requisitos de equilibrio los volúmenes de ambas fases deben ser iguales, para evitar efectos de capacitancia se deber reducir el área interfacial a menos un área aproximada de 1.5 cm2 y para evitar efectos de resistencia incompensada, se debe minimizar la distancia entre los capilares de Luggin y la interfase, ası́ como correr previamente un programa de compensación de caı́da de potencial (Compensación IR) para corregir este defecto [12]. 10 1.2.3. Parámetros Electroquı́micos obtenidos a partir de CV Para que las gráficas obtenidas a partir de Voltamperometrı́a Cı́clica sean útiles en el estudio de las ITIES los potenciales aplicados se referencı́an a una escala [12]. La forma de hacer esto es considerar la suposición termodinámica de que el potencial de transferencia del ion TMA+ es igual a 160 mV para el sistema agua/1,2-dicloroetano y de 35 mV para el sistema agua/nitrobenceno [15]. En cualquier caso el potencial formal de transferencia del ion de estudio se calcula de la siguiente manera: 1/2 ◦ w ◦ w ∆w o φi = ∆o φTMA+ + ∆o φi 1/2 − ∆w o φTMA+ (1.8) Esta expresión es válida solo para la transferencia de un ion de carga z = 1. Los potenciales de media onda se calculan de la siguiente manera a partir del voltamperograma: 1/2 ∆w o φi = f wd ∆w o φi φp ∆w − o 2 (1.9) El primer término de la derecha corresponde al potencial de pico catódico y el segundo término corresponde a la separación de picos en direcciones opuestas de barrido. Para la determinación de concentraciones en la fase acuosa o de los coeficientes de difusión se utiliza la ecuación de Randles-Sevcik (ecuación 1.6) con n = 1 y z = 1. A partir de la intensidad del pico, podemos entonces determinar valores experimentales de variables desconocidas, como por ejemplo el coeficiente de difusión si se cuenta con la concentración de la especie en solución o viceversa [16]. 1.2.4. Parámetros Termodinámicos obtenidos a partir de CV Los parámetros electroquı́micos obtenidos a partir de la información contenida en los voltamperogramas tienen un significado termodinámico también. Al considerar la ecuación 1.4 se observa que la energı́a libre de transferencia está relacionada con el potencial aplicado en la ITIES [14]. El potencial de media onda, corresponde al potencial en el cual las actividades del ion en ambas fases son iguales, además la 11 ecuación 1.8 proporciona la forma de transformar la escala arbitraria medida en una escala absoluta; en esta transformación se asume también que el potencial de onda media es equivalente al potencial formal de transferencia, entonces: ◦ ∆w o φi = ∆◦tr Gw→o i zi F (1.10) La energı́a libre de transferencia se puede interpretar también como la diferencia en energı́as de solvatación del ion [6] en el ambiente acuoso y el ambiente orgánico. Un parámetro adicional que se puede obtener a partir del potencial formal de transferencia es el coeficiente de partición del ion, que proporciona una idea de la distribución de la especie en ambas fases: log Pi = zi F ∆◦tr Gw→o i ∆w φ + RT ln 10 o zi F (1.11) donde se define al coeficiente de partición como: coi log Pi = log w ci ! (1.12) La obtención de parámetros termodinámicos es útil en el análisis de la actividad de sustancias (en este caso, sustancias ionizables o iones per se) en diferentes sistemas, por ejemplo, en los modelos de biodisponibilidad de fármacos [8]. 1.3. Descripción de la técnica de SWV La Voltamperometrı́a de Onda Cuadrada (SWV por las siglas en inglés de Square Wave Voltammetry) es una técnica electroquı́mica de pulsos, es decir, a diferencia de la Voltemperometrı́a cı́clica que sigue una programación lineal de potencial, SWV sigue una programación escalonada caracterizada por la altura del escalón y una frecuencia entre mediciones; al hablar de mediciones se enfatiza una propiedad de la técnica, que es la de muestrear la corriente determinada a un potencial tanto en un sentido como otro (FWD y REV). La técnica forma parte una familia de métodos 12 electroquı́micos que originalmente fueron diseñados para el electrodo de gota de mercurio, como la Polarografı́a de Pulso Diferencial, sin embargo, tiene la caracterı́stica de llevarse a cabo en un electrodo metálico estacionario. Esto último le permite tener la supresión de fondo y sensibilidad de la Voltamperometrı́a de Pulso Diferencial, el valor diagnóstico de la Voltamperometrı́a de Pulso Normal y la habilidad para interrogar al sistema en una forma más versátil que la Voltamperometrı́a de Pulso Reverso [13]. 1.3.1. Fundamentos electroquı́micos de la técnica La programación de pulsos en SWV presentada en la figura 1.5 muestra que hay más variables ajustables a considerar que en el caso de CV. La técnica contempla una rampa de potencial representada por el aumento del escalón ∆Es , un pulso de potencial representado por ∆Ep , un tiempo entre mediciones tp (o bien su inverso, la frecuencia f ) y dos mediciones, una al inicio del pulso (FWD) y otra al final (REV). Figura 1.5: Programación de pulsos en SWV En la figura 1.6 se presenta un voltamperograma tı́pico para una reacción reversible [17] obtenido a partir de SWV. Este presenta tres componentes: la corriente 13 muestreada en dirección FWD, Ψf , la corriente muestreada en dirección REV, Ψb y la diferencia de estas corrientes o corriente neta, Ψnet = Ψf − Ψb . Se puede notar de inmediato que la corriente neta se presenta como una sola onda con un pico bien definido. La intensidad de la respuesta está modelada de la siguiente manera [13]: ∆Ip = nF AD1/2 c∗ f 1/2 ∆Ψnet (1.13) La corriente de pico depende de Ψnet que es un parámetro adimensional definido por Figura 1.6: Voltamperograma de SWV mostrando componentes de la corriente neta. Editado de referencia [17]. la altura del escalón y la del pulso de potencial. La Tabla 1.1 muestra los valores para la corriente adimensional en función de la programación del método. Las condiciones tı́picas de operación para experimentos analı́ticos es mantener el pulso de potencial a 50 mV y la altura del escalón a 10 mV [13]. 1.3.2. Aplicaciones y ventajas de SWV La técnica de SWV presenta ventajas notorias sobre otras técnicas más tradicionales como la voltamperometrı́a cı́clica. Una de las ventajas principales es la capacidad de la técnica para suprimir corrientes de fondo, esto se logra mediante la operación aritmética al restar las corrientes F W D y REV . Por otra parte, la misma operación aritmética permite la obtención de un solo pico máximo. El potencial al que 14 Tabla 1.1: Corrientes de pico adimensionales Ψnet para SWV n∆Ep /n∆Es 2 mV 4 mV 6 mV 0 mV 0.0084 0.0160 0.0230 5 mV 0.1043 0.1116 0.1183 10 mV 0.1984 0.2053 0.2118 20 mV 0.3744 0.3805 0.3863 50 mV 0.7467 0.7499 0.7533 ocurre la corriente de pico es virtualmente el potencial de media onda, lo cual es de gran importancia pues simplifica notablemente el análisis de un fenómeno reversible. Si bien para un proceso reversible la Voltamperometrı́a cı́clica deberı́a ser suficiente para caracterizarlo [14], la facilidad de lectura del Voltamperograma en SWV, la definición de las curvas, la existencia de un solo pico máximo al potencial de media onda y la presentación de los resultados tanto en términos de IREV , IF W D como de Ip efectuada por la mayorı́a de los programas computacionales diseñados para la implementación de la técnica la hacen una herramienta poderosa para aplicaciones analı́ticas; más aún, la supresión de las corrientes de fondo permite una mejor evaluación de las respuestas teóricas [13]. Un factor adicional a la técnica es que se puede obtener la gran cantidad de información mencionada en un tiempo mucho más corto que en CV, ya que la mayorı́a de los análisis tienen una duración de algunos segundos. 1.3.3. Potencial uso de las ITIES con propósitos analı́ticos La aplicación de la técnica de SWV a las ITIES ofrece ventajas diversas si se considera el aspecto analı́tico. En sı́, el control electroquı́mico de la interfase ofrece la oportunidad de detectar solamente iones, con lo cual se suprimen contaminaciones de carga neutra. Adicionalmente esperarı́amos que el transporte de los iones de interés 15 con las caracterı́sticas hidrofı́licas e hidrofóbicas se diera en la región intermedia de la ventana de potencial, con lo cual descartan la observación de señales correspondientes a casos extremos como la mayorı́a de los iones metálicos. La aplicación de la técnica de SWV deberı́a ofrecer mediciones más claras y más fáciles de interpretar que las obtenidas por CV, además de ofrecer mayor sensibilidad y capacidad de diferenciación en el transporte de más de una sustancia en virtud de la caracterı́stica de un solo pico de la respuesta. Las caracterı́sticas descritas en la sección 1.3.2 aplican bien al sistema, dado que se trata con un sistema reversible. La obtención de parámetros como la concentración en solución o los coeficientes de difusión se pueden determinar considerando la respuesta teórica del sistema; asimismo, se puede clasificar a las sustancias de acuerdo a su potencial de pico. Recientemente se ha reportado el uso de SWV en la transferencia de iones plata(I) a través de la interfase agua/1,2-dicloroetano con fines analı́ticos, con un lı́mite de detección de 1.4 µM y pocas interferencias [18]. 1.4. Análisis de sales de Amonio Cuaternario Las sales de amonio cuaternario son compuestos orgánicos iónicos que consisten en un átomo central de nitrógeno con cuatro sustituyentes, mismos que pueden ser hidrógeno, alifáticos o aromáticos y un anión de cualquier ı́ndole. Estas se pueden obtener a través de una sustitución exhaustiva sobre el nitrógeno para dar una sal de amonio cuaternario o bien mediante la halohidratación de una amina primaria, secundaria o terciaria. La estructura general del catión de amonio se presenta en la figura 1.7. La geometrı́a del catión es cercana a la tetraédrica. 16 Figura 1.7: Geometrı́a molecular de un catión de amonio cuaternario. 1.4.1. Importancia de su determinación La determinación cualitativa y cuantitativa de sales de amonio es de gran importancia en las industrias farmacéutica, alimentaria, ası́ como la dedicada a los productos higiénicos y a la producción de envases. Sus usos principales se derivan de sus propiedades antisépticas y tensoactivas [19]. Se hace uso de su función antiséptica en productos sanitarios para granjas, hospitales y otros lugares comunes, también se usan como sanitizantes no oxidativos para la limpieza del agua en albercas, reservas de aguas industriales y de granjas y aguas residuales de hospitales. Son de utilidad asimismo en medicamentos para tratar infecciones del ojo, boca y garganta, ası́ como para uso externo [20]. Su función tensoactiva las hace adecuadas para su uso en la limpieza de botellas de vidrio y plástico como detergentes catiónicos [21], asimismo, se utilizan para conferirle propiedades antiestáticas a ciertas telas, como suavizantes en textiles y productos de papel, en los enjuagues para el cabello y como dispersantes para pigmentos [20]. Debido a las caracterı́sticas germicidas de las sales de amonio, es necesario tener un control cuantitativo sobre su presencia en las aguas residuales [20] y en los productos antes descritos por cuestiones ambientales y de consumo humano [21]. Asimismo, dada su importancia farmacológica, se requiere tener un control estricto sobre su pureza y en un ámbito relacionado, sobre su concentración en fluidos y tejidos con fines medicinales; estas últimas representan un problema más complejo por su presencia en mezclas difı́ciles de analizar [22]. 17 1.4.2. Técnicas analı́ticas existentes para su determinación La cuantificación industrial de aminas y de sales de amonio se realiza aún en términos del nitrógeno total contenido en una muestra, razón por la que se deja a un lado la determinación cualitativa de las especies de interés. Existen métodos de muestreo rápido para detectar residuos de lavado en envases de plástico y vidrio basados en éste principio; éstos se valen de la derivatización de las aminas y sus sales en compuestos volátiles como el óxido de nitrógeno (NO) que se hace reaccionar para dar una reacción quimiluminiscente que puede ser detectada fotométricamente [21]. La determinación de aminas cuaternarias en muestras de alimentos y en fármacos fue explorada en sus inicios por métodos como la cromatografı́a en capa delgada y en papel [19]. La determinación de aminas en estos casos se basa en una reacción colorida con azul de bromofenol o el reactivo de Dragendorff, sin embargo, es claro que una distinción entre compuestos es complicada, más aún, la cuantificación mediante técnicas colorimétricas es sólo aplicable con algunos complejos coloridos formados. Los métodos más novedosos de determinación y cuantificación de aminas y de sales de amonio se basan en cromatografı́a de gases acoplada a espectrometrı́a de masas [22]. La técnica de ionización de superficie (SI) en el espectrómetro de masas ha probado ser adecuada para la determinación de aminas primarias, secundarias y terciarias, llegando a lı́mites de detección del orden de 10−14 g/s. Las aminas cuaternarias se descomponen de acuerdo a una degradación de Hoffman en dicho sistema, sin embargo, la detección de la amina terciaria correspondiente a ésta reacción es posible y eficiente. La técnica ha sido desarrollada teniendo en mente aplicaciones donde la sensibilidad es muy importante, como el caso de mezclas complejas, sin embargo requiere un equipo sofisticado y cuyo mantenimiento es difı́cil. La mayor simplicidad de una técnica electroquı́mica como la propuesta en el presente escrito es ventajosa, aunque presenta el inconveniente de tener lı́mites de detección de orden superior, 10−5 M para CV y 10−6 M para SWV [18], aunque bien su aplicación depende del tipo de muestras para el que esté diseñado el método. 18 1.4.3. Comportamiento de las sales de Amonio en las ITIES El estudio de la transferencia de cationes de sales de amonio cuaternarias y de derivados protonados de aminas ha sido extensamente explorado principalmente por el interés farmacológico que dichas sustancias presentan [9],[23]. Existen asimismo estudios muy detallados de sus propiedades termodinámicas [24] e inclusive de sus efectos sobre las ITIES (como cambios en la tensión superficial en la interfase) dado que algunas de estas sales son utilizadas como electrolitos de soporte para la fase orgánica [25]. Los estudios antes mencionados se centran principalmente en la determinación de coeficientes de partición, lo que es de mayor importancia en farmacologı́a, dejando de lado los aspectos analı́ticos en los cuales el presente estudio se enfoca. La Tabla 1.2 y la Figura 1.8 muestran resultados selectos publicados para cationes de amonio cuaternario. Tabla 1.2: Potenciales de Transferencia y Coeficientes de Partición para Aminas Cuaternarias selectas en el sistema 1-2 Dicloroetano ∆φ◦i / mV log Pi◦ Sustancia Tetra N-pentilamonio −361 6.1 Propantelina −218 3.7 1-Etilquinolina −5 −0.2 Tetra N-metilamonio 160 −2.7 Carbamoilcolina 210 −3.5 Los datos en la Tabla 1.2 muestran que la presencia de grupos hidrofı́licos o una menor oclusión de la carga desplaza el potencial de transferencia hacia valores más positivos, es decir, la energı́a libre de transferencia es más positiva, y por lo tanto, el proceso menos favorable (Ecuación 1.10). 19 Figura 1.8: Estructura de los cationes presentados en la Tabla 1.2. La obtención de parámetros discernibles y cuantificables a través de la combinación de técnicas electroquı́micas (CV y SWV) aplicadas a las ITIES ofrece un área de oportunidad de exploración de gran interés para la ciencia de interfases y en nuestro caso dan pie a la propuesta de desarrollar un método analı́tico para la determinación y cuantificación de sales de amonio en muestras de interés cientı́fico y comercial. Por las razones antes expuestas, el propósito del presente proyecto es evaluar la transferencia simple de iones de amonio cuaternario a través de la interfase polarizable formada entre dos electrolitos inmiscibles a través de las técnicas de Voltamperometrı́a Cı́clica y Voltamperometrı́a de Onda Cuadrada con la intención de desarrollar un método analı́tico cualitativo y cuantitativo para estos. El desarrollo de dicho método contempla la obtención de parámetros electroquı́micos como el potencial de transferencia de dichos iones y el estudio de la correspondencia de la respuesta voltamperométrica con los modelos teóricos, ası́ como la obtención y correlación de la información termodinámica (vbg. la energı́a libre de transferencia) con su estructura y naturaleza quı́mica. La finalidad de esta metodologı́a es adquirir un conocimiento profundo de dichos sistemas que permita identificar las variables crı́ticas y condiciones óptimas para la implementación de las técnicas electroquı́micas aplicadas a la interfase como método analı́tico. 20 Capı́tulo 2 Desarrollo Experimental El presente proyecto tiene como antecedente el trabajo realizado desde Enero de 2004 a cargo del Lic.Vı́ctor Agmo que se desarrolló alrededor de la implementación de la celda electroquı́mica y las primeras mediciones por Voltamperometrı́a Cı́clica para la transferencia de Clorhidrato de Berberina y Cloruro de Colina a través de la interfase agua/nitrobenceno utilizando como sal de soporte orgánica al Tetrafenilborato de Tetrabutilamonio (TBATPB). A partir de Junio de 2004 inicia la segunda etapa del mismo y es donde la presente tesis comienza. 2.1. Preparativos La disposición adecuada y cuidadosa de cada uno de los componentes de la celda electroquı́mica es el punto de partida para la obtención de resultados apropiados y reproducibles en un sistema electroquı́mico. Los preparativos para la realización de un experimento de transferencia de iones a través de una ITIES contempla al menos las siguientes etapas: Sı́ntesis de sales de soporte y preparación de fases, elaboración de electrodos, disposción de la celda electroquı́mica y preparación de las muestras de trabajo. En las secciones siguientes se expone cada uno de estas etapas. 21 El sistema electo para la experimentación queda representado en el siguiente diagrama: Ag|AgCl|BTPPAClAq (7mM), LiClAq (3mM)|BTPPATPBNB (5mM)kLiClAq (10mM), M+ |AgCl|Ag (2.1) Los subı́ndices Aq y NB indican la fase Acuosa y de Nitrobenceno respectivamente. Cada barra vertical indica una interfase y la barra vertical doble la interfase lı́quido-lı́quido sobre la que se centra el estudio. Se puede notar que no es la única interfase lı́quido-lı́quido ya que otra de este tipo se debe utilizar para generar el electrodo de referencia de la fase orgánica, mismo que es un electrodo selectivo para el catión Bis(trifenil)-fosforanilidénamonio (BTPPA+ ). En la fase acuosa no es necesario tener un electrodo selectivo ya que el cloruro de litio en conjunto con el electrodo de Ag/AgCl genera la referencia. Se procura, sin embargo, mantener la misma concentración de iones cloruro en ambos electrodos. La concentración 7mM del BTPPACl en agua está muy cerca de su saturación a temperatura ambiente. La concentración de BTPPATPB en la fase orgánica se eligió en base a la estabilidad que presenta el sistema cuando se trabaja con 5mM de dicha sal de soporte: en general, concentraciones más bajas generaban una alta resistencia de la fase orgánica y concentraciones más altas generaban dificultades instrumentales para controlar el potencial del sistema (mismas que conllevan a la descomposición quı́mica de los solventes). 2.1.1. Preparación de sal de soporte orgánica y fases Como sal de soporte orgánica se eligió el Tetrafenilborato de Bis(trifenil)fosforanilidénamonio (BTPPATPB) ya que se contaba con los reactivos necesarios en el laboratorio para producirla y está reportado [12],[15],[16] que es una de las sales de soporte más adecuadas por permitir una mayor ventana de potencial que otras sales como el Tetrafenilborato de Tetrabutilamonio (TBATPB). La estructura de el BTPPATPB se presenta en la figura 2.1. La sı́ntesis del BTPPATPB consiste en una reacción de precipitación a partir del cloruro de Bis(trifenil)-fosforanilidénamo22 Figura 2.1: Estructura del BTPPATPB. nio ydel Tetrafenilborato de sodio en un medio acuoso [16]. Cantidades equimolares de NaTPB y de BTPPACl se disuelven por separado en la menor cantidad de agua posible, después se mezclan lentamente ambos volúmenes. El resultado es la formación de un precipitado blanco. El precipitado se filtra en frı́o y luego se redisuelve en la mı́nima cantidad de acetona. La disolución de acetona se refluye durante 3 horas, se filtra en caliente, se recoge el precipitado del filtro y se deja secar. Para el periodo de experimentación, se planteó la sı́ntesis de 6 g de sal de soporte. Se partió de una cantidad equimolar de reactivos, aproximadamente 6.5 mmol que significan 2.2329 g NaTPB (PM=342.23 g/mol) y 3.7669 g de BTPPACl (PM=474.05 g/mol). Se obtuvo 4.6024 g de la sal que con un peso molecular de 857.78 g/mol implica un rendimiento de 76.7 %. 2.1.2. Elaboración de electrodos de trabajo Los contraelectrodos para la fase acuosa y orgánica consistieron en alambres de platino (o bien alambres con un extremo libre en forma de disco) de aproximadamente 5 cm de largo insertados dentro de un capilar de vidrio y sellados de manera que sólamente una porción de 3 cm sobresaliera. Para facilitar el contacto entre el cable conductor y el platino, evitando a su vez la necesidad de soldar platino a cobre, se realizó el contacto óhmico con mercurio. Una vez que el platino se sellaba por un extremo del capilar de vidrio, se introducı́a la cantidad necesaria de mercurio 23 para llenar la mitad del capilar; enseguida, se introducı́a el cable conductor de cobre debidamente lijado y limpio y finalmente, a través del extremo superior (abierto) del capilar, se introducı́an algunas gotas de naftaleno lı́quido caliente. Al enfriarse, el naftaleno solidifica, por lo que es necesario calentar los extremos del capilar en la flama de un mechero para que éste resbalara por las paredes y ası́ obtener un sellado adecuado. El sellado con naftaleno es necesario por cuestiones de seguridad, debido a la toxicidad y la alta presión de vapor del mercurio. Los electrodos obtenidos se muestran en la figura 2.2. Figura 2.2: Electrodos de Trabajo de Platino. A)Conexiones hembra, B)Alambre de cobre dentro de capilar de vidrio, C) Sello de naftaleno, D)Mercurio en contacto con alambre de cobre y alambre de platino, E)Alambre de platino en contacto con Mercurio y a la vez sellado en el extremo del capilar de vidrio. 2.1.3. Elaboración de electrodos de referencia Para la disposición de una celda electroquı́mica para el estudio de las ITIES se deben utilizar dos electrodos de referencia, uno respectivo para cada fase. En la fase acuosa se utiliza un electrodo de referencia Ag/AgCl con el antecedente de que la sal de soporte acuosa contiene el anión cloruro. En la fase orgánica se debe usar un electrodo selectivo a alguno de los iones de soporte; esta tarea no es sencilla y generalmente 24 se opta por establecer un electrodo selectivo basado en el electrodo Ag/AgCl. El electrodo de la fase orgánica queda entonces conformado por una interfase Ag/AgCl en contacto con una solución de cloruros y de alguno de los iones presentes en la sal de soporte orgánica (en el caso de este estudio BTPPA+ ); esta solución genera a su vez una interfase con la fase orgánica que contiene la sal de soporte. Finalmente el control del potencial a través del capilar de Luggin se da gracias a esta interfase entre la referencia acuosa y la fase orgánica, misma que permite el paso de iones BTPPA+ para sensar el potencial en la ITIES donde se lleva a cabo la transferencia del ion de estudio. La fabricación de electrodos de referencia Ag/AgCl resultó de gran importancia para el desarrollo experimental; nuevos electrodos se generaban al inicio de cada sesión experimental para garantizar la estabilidad de la aplicación y medición del potencial en la interfase. El buen comportamiento de los electrodos de referencia es crucial para evitar medidas erróneas de potencial ası́ como para garantizar la estabilidad del sistema que se trabaja, con especial énfasis en que un sistema como el del presente estudio puede desestabilizarse fácilmente. La fabricación de un electrodo de referencia Ag/AgCl se basa en la siguiente reacción: Ag(s) + Cl− (aq) −→ AgCl(s) + ē (2.2) Para llevar a cabo esta reacción, se partió de alambres de plata soldados previamente a un contacto hembra (para conectarse con facilidad a la caja del electrómetro). El alambre se conecta a la terminal positiva de un arreglo en serie de cuatro pilas AA cuya diferencia de potencial fluctuaba entre 5 V y 6 V. En la terminal negativa se utilizó un contraelectrodo de platino. Ambos electrodos se sumergen en 100 ml de una solución 0.1 M de KCl y tres gotas de HCl concentrado para reducir la solubilidad del cloruro de plata. Se deja un tiempo de electrólisis de entre 20 y 30 segundos. Una vez obtenidos los electrodos, éstos se guardaban en una caja de petri aislada forrada con papel aluminio para evitar la descomposición del cloruro de plata a causa de la luz. 25 La obtención exitosa de los electrodos de referencia se puede anticipar de manera rápida por inspección directa con ayuda de un estereoscopio de la superficie donde se ha realizado el depósito. Un electrodo adecuado presenta un recubrimiento blanco uniforme, sin puntas metálicas. Durante el proceso de electrólisis no se debe observar el desprendimiento de burbujas sobre el electrodo de plata, de lo contrario, es un indicativo de que la reacción sobre la plata es incompleta a causa de una impureza. Esta última observación fue recurrente en los alambres de plata que se reusaban sin haber sido limpiados adecuadamente del depósito anterior de AgCl. Para reusar un electrodo que ya ha sido sometido al depósito de AgCl (como generalmente se hizo, reusando el alambre de plata de 4 a 5 veces), se debe seguir un procedimiento de limpieza y reducción de la superficie. El electrodo usado de Ag/AgCl se lija con papel de graduación 200 o 400 hasta retirar la capa depositada. A continuación se limpia la superficie con agua. El extremo del alambre de plata se sumerge después en una solución aproximadamente 0.5M de NaHCO3 a punto de ebullición. El contacto hembra se conecta con un cable caimán a una lámina de Zinc lijada y dicha lámina se sumerge en el mismo baño. Se deja reposar el circuito aproximadamente 5 minutos. Una vez transcurrido este tiempo, se puede lavar el electrodo con agua destilada seguida de acetona. El electrodo está listo para depositar de nuevo una capa de AgCl. La reducción con Zinc consiste en la siguiente reacción: 2AgCl(s) + Zn(s) −→ 2Ag(s) + ZnCl2(aq) (2.3) La reacción tiene una energı́a libre de Gibbs a 95◦ C de −99 kJ/mol, prefiriéndose esta temperatura por causas cinéticas. La función de este tratamiento es remover por completo cúmulos de plata oxidada sobre la superficie que eviten una deposición uniforme. Un aspecto adicional a considerar en cuanto a la eficiencia de la deposición, es que el baño de cloruro de potasio debe ser cambiado aproximadamente cada 20 o 30 deposiciones puesto que una vez que la concentración de cloruro en dicha solución es reducida los resultados son inadecuados. Los electrodos fabricados adecuadamente deben ser como los de la Figura 2.3. 26 Figura 2.3: Electrodos de Referencia Ag/AgCl, A)Alambre de plata recubierto de cloruro de plata, B)Conexión hembra. 2.1.4. Disposición de la celda electroquı́mica Las mediciones se llevaron a cabo en una celda electroquı́mica acondicionada con tres orificios para colocar los dos electrodos de referencia y los contraelectrodos de cada fase. En la primer etapa de la investigación se utilizó una celda electroquı́mica de un área interfacial 1.17 cm2 , para la segunda etapa se contaba ya con una celda de 0.49 cm2 . La búsqueda de la reducción del área interfacial corresponde a la hipótesis de que una interfase más pequeña debe ofrecer una menor corriente capacitiva y un mejor control de las condiciones experimentales (por ejemplo, una medición más uniforme de la corriente faradáica y del potencial a través de los capilares de Luggin). La Figura 2.4 muestra la celda de vidrio utilizada en la mayor parte de la experimentación. La celda se montó en una base de aluminio para garantizar la estabilidad mecánica del sistema. Debido a la diferencia de densidades de los solventes, la fase orgánica quedó en la parte inferior de la celda, lo cual es ventajoso debido a que se previene la evaporación del solvente orgánico. 27 Figura 2.4: Celda de vidrio con área interfacial de 0.51 cm2 , (A) Brazo lateral para la la referencia orgánica, (B) Tubo principal para la introducción de contraelectrodos, (C) Brazo lateral para referencia acuosa. El mecanismo de llenado siguiendo la notación de la Figura 2.4 se describe a continuación. Todas las soluciones de trabajo fueron introducidas con el uso de jeringas desechables de plástico de 3 ml de capacidad acondicionadas con una aguja hipodérmica de 5 cm de largo. La limpieza de las jeringas se realizaba regularmente tomando volúmenes de agua destilada, agitando y desechando el residuo hasta no observar turbiedad en el agua desechada. Los volúmenes de disolución orgánica y de disolución acuosa fueron 1.5 ml para la celda pequeña y 4.1 ml para la celda grande. 1. Se introduce la jeringa con la disolución orgánica en el brazo (A) y se inyecta lentamente evitando que un impulso demasiado enérgico del émbolo cause que parte de ésta se transfiera al capilar de Luggin superior (con los volúmenes sugeridos, el nivel de la fase no superará la mitad de la distancia entre los capilares de Luggin). 2. Se cubren los orificios de (B) y (C) con parafilm y se sellan de manera que no entre aire al sistema. Se puede presionar ligeramente el orificio de (B) para elevar un poco el nivel de nitrobenceno en el tubo (A). 28 3. Se introduce la jeringa con la disolución de referencia orgánica en el tubo (A) y se inyecta una pequeña cantidad de ésta (aproximadamente 0.1 ml) con cuidado de no introducir burbujas de aire y de no hacerlo con suciente fuerza como para perturbar la fase orgánica. 4. Se cubre el orificio (A) con parafilm para sellarlo y se destapan los orificios (B) y (C). 5. Se introduce la jeringa con disolución acuosa y se vacı́a la disolución lentamente permitiendo que se establezca la interfase. 6. Se destapa el orificio (A) y se deja equilibrar el sistema. La interfase debe quedar a la mitad de los capilares de Luggin. 7. Para garantizar la planaridad de la interfase se pueden tomar pequeños volúmenes de la disolución de referencia orgánica con la jeringa correspondiente. Una vez llenada la celda se puede proceder a la colocación de los electrodos. Se inicia haciendo las conexiones a la caja del electrómetro como se muestra en la Figura 2.5. Una vez hechas las conexiones, los electrodos se colocan en los los orificios correspondientes. En el caso de los brazos para las referencias basta con sumergir la el electrodo de Ag/AgCl (cuidado de no sumergir la partes sin depósito o que en el caso de la referencia orgánica el electrodo toque la interfase con la fase orgánica). Para el tubo principal (orificio B) se colocan los contraelectrodos de platino apoyados mutuamente de manera que se puedan acomodar y queden fijos. Se sugiere que ambos contraelectrodos sean equidistantes a la interfase y que queden aproximadamente a la altura de los capilares de Luggin puesto que se observó que éste arreglo favorece un regreso más rápido al potencial de circuito abierto al realizar corridas consecutivas. El aspecto final de la celda se muestra en la Figura 2.6. En la configuración de electrodos 29 Figura 2.5: Configuración de las conexiones en la caja del electrómetro. (A) Hacia la referencia orgánica, (B1) Hacia el contraelectrodo orgánico, (B2) Hacia el contraelectrodo acuoso, (C) Hacia la referencia acuosa. descrita, el potencial a través de la interfase obedece la ecuación 1.7; una polarización positiva implica entonces hacer la fase acuosa más positiva con respecto a la orgánica. A mitad del periodo experimental se presentaron algunas interferencias electrónicas en las mediciones. Dichas interferencias pueden provenir de ruido en la red eléctrica o deberse al uso de motores en regiones cercanas al laboratorio; Este inconveniente se resolvió construyendo una jaula de Faraday para aislar el sistema. El funcionamiento de la caja de Faraday no se probó estrictamente, sin embargo, una buena guı́a fue introducir un teléfono celular dentro de ella. Como era esperado, la señal de recepción del teléfono disminuyó a un mı́nimo, lo que es una indicación del buen funcionamiento de la jaula, más aún, no se observó interferencia posterior de fuentes desconocidas (aunque sı́ la hubo posteriormente cuando se introdujo un circulador termoestable para el arreglo experimental necesario para la transferencia a distintas temperaturas). La Figura 2.7 muestra el sitema final. 30 Figura 2.6: Aspecto final de la celda electroquı́mica una vez llenada y colocados los electrodos. 2.1.5. Arreglo experimental para variación de temperaturas Con la finalidad de estudiar la transferencia de iones a distintas temperaturas, se incorporó al sistema descrito un sistema de control de temperatura. Se construyó un recipiente con una entrada inferior y una superior para introducir una mezcla 5:1 de agua:glicerina a través de un recirculador provisto por un baño de temperatura constante. El baño de temperatura constante fue un dispositivo MGW-Lauda R18. Se trabajaron temperaturas entre 5o C y 70o C. La celda electroquı́mica fue introducida en su totalidad en el recipiente externo. El recipiente estuvo hecho de vidrio, se recubrió con 3 capas de papel aluminio comercial y encima una capa de poliuretano. El recipiente cubierto se envolvió en cinta de aislar negra. La celda electroquı́mica se montó sobre un tapón monohorado para garantizar su estabilidad mecánica. El sistema tuvo que ser elevado para disminuı́r el flujo de agua proveniente del recirculador; para este propósito también se estableció un sistema de mangueras en paralelo para bifurcar el flujo (una entrada hacia el recipiente de vidrio y la otra entrada hacia el depósito principal de lı́quido en el baño). El arreglo se muestra en la Figura 2.8. Debido a complicaciones instrumentales, no se pudo utilizar el potenciostato mientras el recirculador permaneciera encendido, por lo que el sistema se estabilizaba durante 31 Figura 2.7: Celda dentro de Jaula de Faraday. Se debe tener especial precaución en no perturbar el arreglo de electrodos al abrir y cerrar la jaula. un mı́nimo de 5 minutos a la temperatura deseada y posteriormente se apagaba el recirculador, se prendı́a el potenciostato, se medı́a y se apagaba. El proceso se repetı́a hasta obtener las mediciones deseadas. Desgraciadamente tampoco se pudo utilizar un termómetro digital, razón por la cual se incorporó un termómetro de mercurio con precisión de 1o C. La complicación instrumental se atribuye a la interferencia de señales electrónicas entre los aparatos y el potenciostato; dicha señal inducı́a una corriente en el rango de nA y causaba oscilaciones fuertes en el potencial de la celda. 2.1.6. Preparación de disoluciones En esta sección consideraremos la preparación de tres tipos de disoluciones: la disolución de referencia para el electrodo selectivo a BTPPACl, la disolución para la fase orgánica y la disolución para la fase acuosa, que adicionalmente contiene la sal de estudio. Todas las muestras acuosas se prepararon con agua desionizada 18.2MΩ/cm obtenida de un desionizador EasyPure II. Preparación de disolución de referencia orgánica: La solución para el electrodo de referencia orgánica se preparó disolviendo BTPPACl en agua, la concentración electa 32 Figura 2.8: Dispositivo para mediciones a distintas temperaturas, (A) Recirculador con bifurcación de flujo integrado, (B) Recipiente de vidrio con entrada y salida para alojar en su interior a la celda electroquı́mica. fue de 5mM . Se utilizó BTPPACl (Aldrich 98 %) como se recibió del proveedor. La cinética de disolución de esta sal en agua es lenta, por lo que durante el proceso se debe agitar fuertemente con ayuda de un agitador de vidrio; el proceso de disolución a temperatura ambiente toma aproximadamente 20 minutos y generalmente la disolución resultante genera algo de espuma que después de un tiempo desaparece. La solución resultante se saturó con una gota de nitrobenceno (para aproximadamente 25 ml de la disolución). El equilibrio previo con nitrobenceno se debe hacer con la cantidad mı́nima de este debido a que un exceso causa en general un enturbiamiento de la disolución. Preparación de la disolución para la fase orgánica: Se utilizó nitrobenceno (Aldrich 99 %) sin posterior proceso de purificación. Para la obtención de una disolución 5mM de BTPPATPB se disolvı́an 0.1047 g de la sal en 25 ml de nitrobenceno, el proceso de disolución en general era rápido. La disolución enseguida era saturada con 1 gota de agua y se agitaba fuertemente. Se observó que durante periodos prolongados de tiempo y en exposición a la luz, el color de la disolución cambiaba de un amarillo claro a un amarillo intenso, razón por la cual el matraz se cubrió de papel aluminio. 33 Preparación de la disolución acuosa: La disolución acuosa contenı́a la sal de soporte acuosa y la sal correspondiente al cloruro del ión de estudio. Se eligió como sal de soporte al Cloruro de Litio (Baker 99 %) en una concentración 10mM . Un procedimiento que simplifica notablemente la preparación de muestras es la preparación de una solución stock 100mM de LiCl a partir de la cual se pueden preparar las disoluciones 10mM . Una ventaja adicional de la preparación de la solución stock inicial con concentración mayor es minimizar el error en la preparación ya que debido al bajo peso molecular del LiCl, la cantidad requerida de sal era muy baja (para 25 ml), adicionalmente, el LiCl es una sal muy higroscópica y pesar pequeñas cantidades puede introducir un error considerable. La preparación de las muestras de trabajo se realizó utilizando como base las disoluciones 10mM de LiCl. Se prepararon soluciones de Cloruro de Tetrametilamonio (TMACl, Aldrich 99 %), Cloruro de Colina (CHCl, The Biochemical Coorporation, 98 %), Cloruro de Tetrabutilamonio (TBACl, Aldrich 98 %). Para minimizar el error, igualmente se prepararon soluciones stock 2mM del ion de estudio a partir de las cuales se redujo la concentración a 0.4 mM y finalmente a la concentración deseada a partir de esta. En el caso del TMACl se prepararon disoluciones de concentraciones entre 10 y 400 µM , sin embargo, la transferencia a concentraciones mayores a 200µM se descartó por no presentar utilidad alguna. No se siguió un patrón estricto en cuanto a las concentraciones del ión de estudio, sin embargo, se procuró que la distancia entre cada punto para la construcción de las curvas de calibración fuera de entre 10µM y 20µM . 2.2. Técnicas Experimentales En el presente trabajo se estudió la transferencia de iones a través de las técnicas de Voltamperometrı́a Cı́clica (CV, sección 2.2.2) y Voltamperometrı́a de Onda Cuadrada (SWV, sección 2.2.3), sin embargo, una técnica adicional que se introduce 34 debido a las caracterı́sticas de la celda electroquı́mica (la presencia de un solvente orgánico y de múltiples interfases además de la de interés) es la Compensación IR. A continuación se presenta una descripción de los parámetros utilizados en las técnicas y las recomendaciones generales de uso con respecto al comportamiento que mostró el potenciostato y la interfase computacional. 2.2.1. Compensación IR Debido a la naturaleza de la composición del sistema (que incluye el uso de un electrolito orgánico) se debe de compensar la resistencia que este ofrece. El arreglo experimental utilizado se puede representar mediante un circuito electrónico equivalente de Randles como se muestra en la Figura 2.9 [26]. Por el momento es suficiente discutir el papel de la resistencia de la solución, Rs , sobre el sistema. En el caso del electrolito acuoso, podemos esperar que la resistencia que este presenta al paso de corriente sea mucho menor a la ofrecida por el electrolito orgánico, principalmente debido a que el solvente orgánico (nitrobenceno) tiene una constante dieléctrica menor que la del agua y consecuentemente será menos eficiente en ionizar la sal de soporte (esto implica asimismo una menor conductividad eléctrica per se). La resistencia que ofrece el electrolito orgánico obedece a la ley de Ohm: ∆E = IRs (2.4) En la Ecuación 2.4 ∆E representa una caı́da de potencial e I la corriente que fluye por el sistema, la cual podemos restringir a los procesos de transferencia del ión de estudio y de las sales de soporte. La caı́da de potencial a través de la solución orgánica causa entonces una discrepancia entre el potencial aplicado por el potenciostato y el potencial que realmente existe en la interfase, ésta caı́da es mejor descrita en todo caso como un potencial de error. Para compensar dicho error se introduce entonces un término de compensación que consiste en una sustracción de una cantidad medida experimentalmente por el potenciostato. Esta compensación, sin embargo, no puede llevarse a cabo en su totalidad ya que esto ocasiona a menudo oscilaciones en el 35 sistema electrónico del potenciostato. La idea es compensar tanto como sea posible. La Ecuación 2.5 muestra el efecto de la compensación [27]: EApp = Eint + Eerr − αIRs (2.5) En donde se ha renombrado a ∆E como Eerr y EApp es el potencial deseado que se aplica (y que se mide). Eint es efectivamente el potencial que se establece a través de la interfase y se ha introducido un parámetro α que es un valor numérico entre 0 y 1 que describe el grado de compensación. Como se mencionó con anterioridad, a menudo no se pueden alcanzar valores cercanos al 1 para α, pues esto implicarı́a una compensación total y la consecuente pérdida de estabilidad en el sistema [27]. Figura 2.9: Circuito equivalente de Randles para una ITIES, Rs indica la resistencia incompensada de la solución, Cint la capacitancia de la interfase, ZIT la impedancia resultante de la transferencia iónica y ZB la impedancia resultante de la transferencia de las sales de soporte. El método utilizado en el presente trabajo para realizar la compensación fue el de Positive Feedback . El procedimiento consiste en la aplicación de un pulso de potencial a un valor determinado del potencial de la celda (por ejemplo el potencial de circuito abierto). Debido a la excitación del pulso, una corriente fluye por el sistema. A través del sistema de procesamiento de datos (computadora) se analiza automáticamente el decaimiento de la corriente después del pulso de potencial. El decaimiento de la corriente obedece para el proceso controlado por la resistencia de la solución a un decaimiento exponencial en el cual la constante de tiempo es equivalente a Rs Cint . El sistema entonces puede calcular el valor de Rs [27]. Al realizar el experimento, el sistema compensará el producto αIRs del potencial aplicado, el cual, evidentemente 36 depende de la corriente que fluye por el sistema. Dicha sustracción es discutible ya que el valor de Rs puede variar en el trascurso del experimento y es una fuente de error considerable, sin embargo es la mejor aproximación que se tiene para evitar un error mayor. La tabla 2.1 resume el valor de los parámetros utilizados para la Compensación IR. Las resistencias calculadas tı́picas fueron de entre 200 y 800 Ω para los electrodos de alambre de platino y de entre 700 y 1500 Ω para los electrodos de disco de platino. Dichos valores están influenciados principalmente por el arreglo de los electrodos y de la interfase (la distancia entre los contraelectrodos con la interfase, la posición de la interfase entre los capilares de Luggin y la temperatura del sistema). Es de especial importancia mencionar que el montaje de la celda debe hacerse con cuidado para que el programa de Compensación IR funcione adecuadamente; un problema importante durante la etapa experimental fue que cuando la celda no era montada apropiadamente, el software de la computadora presentaba un error y se cerraba, dejando al potenciostato fijado en el valor de potencial. El software se debı́a abrir de nuevo y ajustar el potencial. Un parámetro denominado Undershoot se posicionaba en un valor de 5 %, este es importante pues determina el grado de oscilación tolerable para el sistema; cuando funciona adecuadamente el software no debe presentar el error mencionado. Durante las mediciones con el programa de Compensación IR el potenciostato era notablemente sensible, por lo que cualquier interferencia electrónica (como el motor de la campana de extracción u otros aparatos cuyas frecuencias interfirieran) causaba un descontrol del potencial y la consecuente degradación de las soluciones, como se muestra en la Figura 2.10. 2.2.2. Voltamperometrı́a Cı́clica (CV) Una descripción de la técnica ya se ha dado en la sección 1.2 y subsecuentes. En este apartado se mencionarán los parámetros de operación y las razones para elegir estos. La Tabla 2.2 muestra las condiciones de operación utilizadas en la mayorı́a de los experimentos. 37 Tabla 2.1: Parámetros utilizados en Compensación IR siguiendo la nomenclatura del Research Electrochemistry Software 4.0 de EG&G. PARÁMETRO VALOR OBSERVACIONES Pulse Height (PH) −10 mV Se puede aumentar a −20 mV Compensation Level (CL) 70 % No elevar a más de 80 % Undershoot (UN) 5% No aumentar ni disminuir Measuring Potential (MP) Voc + 0.02 mV Conveniente no sobrepasar Current Range (CR) 100µA Válida sólo a este CR IR Mode (IR) Measured Entered es para aplicarla Electrode Solid - Reference Electrode User - En general se utilizó un valor de Velocidad de Barrido, ν (Ecuación 1.6), de 10 mV/s por dos razones principales. La primera es que a velocidades mayores se observaba un ligero desplazamiento en el potencial del pico en sentido de polarización positiva (y en su contraparte negativa también). Este no es un comportamiento esperado de acuerdo a lo establecido en la sección 1.2; más aún, la aparición de dos ondas en la polarización negativa o bien de un distanciamiento mayor al esperado entre las corrientes de pico en ambas direcciones de polarización dificultaba la observación del potencial de onda media. La segunda razón es que a velocidades de barrido superiores la corriente capacitiva (3.1.1) aumenta con respecto al término lineal de dicha velocidad, lo cual es indeseable para el tratamiento de la señal a bajas concentraciones del ion de estudio debido a que el empalme con la señal de transferencia llega a ser significativo. A concentraciones mayores una velocidad de barrido alta causa una deformación de la respuesta electroquı́mica y resultados poco confiables. El uso de velocidades de barrido bajas (no mayores a 20 mV/s) es sugerido extensamente [10]. 38 Figura 2.10: Descomposición de las soluciones debida a una oscilación fuerte en el potencial aplicado. Esto sucede cuando se presenta un descontrol instrumental durante un experimento. Una complicación relacionada con el rango de corriente (CR) se presentó debido a que las caracterı́sticas del arreglo experimental (y su interacción con el potenciostato) no permitieron el uso de un rango menor. Un rango de corriente menor implica una mayor sensibilidad de la respuesta al aumentar un decimal en la corriente medida. A bajas concentraciones del analito la corriente de pico no podı́a ser leı́da con precisión puesto que aparecı́a ”achatada”; asimismo esta señal deforme se extendı́a en un rango de potencial apreciable, por lo que la lectura del potencial de pico se veı́a afectada también. Al correr la voltamperometrı́a cı́clica, se debe descartar en general la primer medición. Por esta razón el número de ciclos se incrementaba. Un número razonable de estos es tres, aunque si el sistema no mostraba una variación significativa entre el primer ciclo y el segundo, se podı́a detener al finalizar este. Un aspecto muy importante en las mediciones, es que se debe utilizar un filtro para frecuencias de interferencia (Low Pass) debido al ruido introducido por la lı́nea eléctrica o bien por las lámparas del laboratorio. Durante la experimentación se observó que el filtro adecuado es el de 590 Hz. 39 Tabla 2.2: Parámetros utilizados en CV siguiendo la nomenclatura del Research Electrochemistry Software 4.0 de EG&G. PARÁMETRO VALOR OBSERVACIONES Scan Rate (SR) 10 mV/s Hasta 20 mV/s Equilibrium time (ET) 15 s Se puede reducir a 10 s Purge Time (PT) pass No aplica Initial Potencial (IP) Voc Hasta Voc + 0.02 V Vertex 1 (V1) 0.8 V Conveniente no sobrepasar Vertex 2 (V2) 0.05 V Conveniente no sobrepasar Final Potential (FP) Voc - Number of cycles (NC) 3 A veces con 2 es suficiente Store cycle (SC) 3 2 si aplica IR Mode Entered Una vez corrida la compensación Current Range (CR) 100 µA Funciona sólo en este rango Electrode Solid - Electrode Area (EA) 1 cm2 Por convención Reference Electrode User - Filter 590 Hz Filtra el ruido eléctrico de las lámparas 2.2.3. Voltamperometrı́a de Onda Cuadrada (SWV) Para las mediciones en Voltamperometrı́a de Onda Cuadrada se utilizaron los parámetros mostrados en la Tabla 2.3. Al igual que para CV (Sección 2.2.2), una limitación fue el rango de corriente que se tuvo que fijar. La técnica de SWV es más sensible cuando el pulso de potencial aplicado es cercano a 50 mV como lo muestra la Tabla 1.2. Valores mayores a 20 mV (el signo negativo se usa indistintamente 40 como convención para el arreglo de electrodos utilizado en la experimentación) no se pudieron accesar debido a que en los extremos de la ventana de potencial las componentes de la corriente neta en SWV sobrepasan a menudo el valor tolerable para el rango de corriente (para CR=100µA, el lı́mite es ±120µA), causando un cese en la medición de corriente (respuesta muerta). El valor adecuado para la experimentación, en el que se obtiene una mejor respuesta electroquı́mica sin comprometer la estabilidad del sistema o la medición es de −20 mV. Después de la obtención del voltamperograma de onda cuadrada, se procedı́a a un suavizado de la curva, esto se hacı́a seleccionando la opción Numerical/Smooth en el software de adquisición. En toda la experimentación se utilizó el algoritmo de Sliding Average de grado 5. Tabla 2.3: Parámetros utilizados en SWV siguiendo la nomenclatura del Research Electrochemistry Software 4.0 de EG&G. PARÁMETRO VALOR OBSERVACIONES Pulse Height (PH)* −20 mV Máximo tolerable por el sistema Scan Increment (SI)* 2 mV Determina la densidad de información Frequency (FR)* 10 Hz Aumentarla deforma la respuesta Initial Potential (IP) 0.05 V A veces 0.1 V Final Potential (FP) 0.85 V Conveniente no sobrepasar Equilibrium Time (ET) 15 s A menudo reducido a 7 s IR Mode Entered Una vez corrida la compensación IR Current Range (CR) 100 µA Funciona sólo en este rango Electrode Solid - Electrode Area (EA) 1 cm2 Por convención Reference Electrode User - Filter 590 Hz Filtra el ruido eléctrico de las lámparas *En el desarrollo del texto, y sobre todo para corresponder a las ecuaciones presentadas, las variables tienen la siguiente notación equivalente: P H = ESW , SI = Es y F R = f = τ1 . 41 Capı́tulo 3 Resultados y Discusión En la presente sección se discutirán los resultados más importantes obtenidos durante la experimentación en el orden en que fueron obtenidos. Para facilitar la estructura del presente trabajo la discusión de cada resultado se hará de manera simultánea a su presentación. Los resultados se han estructurado de acuerdo al siguiente esquema: optimización de la calidad de la señal electroquı́mica (sección 3.1), obtención de mediciones con propósitos analı́ticos (Secciones 3.2 y 3.3), evaluación de señales electroquı́micas y transferencias mixtas (Sección 3.4) y finalmente el comportamiento de la transferencia iónica a distintas temperaturas (Sección 3.5). 3.1. Preparativos En esta sección se discutirá la obtención de los parámetros de operación óptimos para cada técnica; dicha decisión se fundamentó principalmente en la reproducción de resultados y en el reconocimiento del origen de las corrientes de fondo, ası́ como la relación entre estas y la corriente producto de la transferencia de un ion a través de la interfase. 42 3.1.1. Comportamiento de las Corrientes de Fondo La interfase entre los dos electrolitos inmiscibles se comporta como un capacitor; esto es evidente a partir del circuito equivalente de Randles presentado en la Figura 2.9. Un detalle importante acerca de la interfase, es que la capacitancia que presenta en general es alta comparada con otros sistemas electroquı́micos, por ejemplo, la interfase metal/acuoso tradicional. En los sistemas tradicionales metal/acuoso dicha capacitancia se interpreta en términos de la teorı́a de la doble capa desarrollada por Helmholtz y mejorada por Gouy y Chapman [14]. La teorı́a de la doble capa propone que en la interfase existe una adsorción de especies cargadas o polarizadas; por ejemplo, en una interfase metal/acuoso, el metal (negativo) inducirá una polarización sobre las moléculas de agua que se adsorberán orientadas preferencialmente sobre los átomos de hidrógeno. Igualmente, los cationes hidratados de una especie electroactiva se adsorberán sobre la superficie del metal. Dado que el fenómeno de adsorción es dependiente del potencial en la interfase, también lo será la capacitancia y por lo tanto, un cambio en el potencial induce un cambio en la carga de la interfase, que se traduce en una corriente que fluye por el sistema. La demanda de carga (y por lo tanto la Corriente capacitiva que presenta el sistema) está definida por el comportamiento de la interfase; para el caso de las ITIES la ausencia de datos precisos sobre la estructura de la interfase hacen difı́cil su estudio [6], sin embargo el primer paso para permitir el tratamiento de datos requerido para las aplicaciones analı́ticas en las que se fundamenta el presente estudio es identificar a las corrientes de fondo con su origen capacitivo. Una manera de identificar el proceso capacitivo es mediante la variación de la velocidad de barrido (en CV) o la frecuencia (en SWV). De la definición de capacitancia observamos: Cd = dQ I = dE ν (3.1) dQ dt (3.2) dado que I= 43 y ν= dE dt (3.3) Cd representa la capacitancia diferencial de la interfase. El hecho de que se trate de una capacitancia diferencial implica por supuesto que su valor será diferente a diferentes valores del potencial, E. Por su parte, ν puede ser interpretada directamente en CV o bien, con su equivalente en SWV, donde la velocidad de barrido será el producto de la altura del escalón por la frecuencia ν = f ∆Es . La Ecuación 3.1 sugiere que graficar la corriente capacitiva contra la velocidad de barrido debe dar una lı́nea recta con una pendiente proporcional a la capacitancia. Esto solo será valido a un potencial definido. Para CV los resultados de este análisis se presentan en las Figuras 3.1 y 3.2. Estos corresponden a la celda grande (área geométrica= 1.17 cm2 ). Es importante la mención que la proporcionalidad de la capacitancia también vendrá dada en función del área de la interfase; su relación es lineal, ası́ que es extrapolable a la celda pequeña. La Figura 3.1 retrata las lı́neas base tı́picas que se observaron en CV. Nótese que Figura 3.1: Corrientes de fondo en CV. Al aumentar la velocidad de barrido se aprecia un aumento en las corrientes de fondo. al realizar el barrido reverso no hay una recuperación total de la corriente, lo que se 44 Figura 3.2: Linealización de la corriente de fondo medida a 0.55 V en CV contra la velocidad de barrido. Se demuestra una tendencia lineal aceptable que confirma el efecto de la capacitancia de la interfase evidencı́a en el signo negativo que predomina. La ventana de potencial que la técnica permite explotar es de aproximadamente 0.6 V que se encuentra entre 0.1 y 0.7 V. En este intervalo se observa que la separación entre las señales a diferentes velocidades de barrido es aproximadamente equidistante, por lo que el análisis hecho en la Figura 3.2 es válido para distintos potenciales en este rango. En efecto, este rango en el que la capacitancia diferencial no cambia apreciablemente es el que se puede explotar para observar la transferencia de un ion de estudio. Para el caso de SWV, se puede variar la frecuencia para evaluar el comportamiento de las corrientes de fondo de una manera equivalente a la ya hecha en CV. El comportamiento de dichas corrientes de fondo es presentado en la Figura 3.3. Se puede llevar a cabo la linealización con respecto a la frecuencia como se muestra en la Figura 3.4. En el caso de SWV observamos un mejor acuerdo entre la variación de las corrientes de fondo contra la frecuencia para un modelo lineal. Un punto discutible 45 Figura 3.3: Variación de las corrientes de fondo en SWV al variar la frecuencia f del barrido, P H=−10 mV en este sentido es que uno de los aspectos positivos de la técnica de SWV es la supresión de componentes no-Faradáicas y componentes capacitivas de la señal neta. Si bien la componente capacitiva no se elimina, el mejor ajuste lineal indica que las componentes no-Faradáicas estan siendo suprimidas. La separación entre las curvas a distintas frecuencias es aproximadamente equidistante, lo cual es un comportamiento acorde a lo ya observado en CV. Ahora bien, un parámetro adicional que se debe tomar en cuenta para el análisis por SWV es la altura del pulso, P H. Si bien en la Ecuación 3.1 no se sugiere directamente su uso, una observación más profunda del fenómeno de transferencia en SWV lo hace más evidente. La onda cuadrada superpuesta sobre la programación lineal de potencial en SWV dará como resultado una variación local (es decir, en cada valor de potencial) de la corriente capacitiva. En tanto esta sea caracterizable, no deberı́a presentar problema alguno. Se puede comparar la pendiente (a decir, la constante de proporcionalidad capacitiva) a diferentes valores de P H como se muestra en la Tabla 3.1. 46 Figura 3.4: Linealización de las corrientes de fondo en SWV al variar la frecuencia f del barrido, P H=−10mV, E=0.55 V La Tabla 3.1 muestra que la selección de valores mayores para P H en las mediciones afecta notablemente la constante de crecimiento de las corrientes de fondo. Las columnas que muestran la relación entre los valores en la corriente de fondo y los valores teóricos de Ψ para los diferentes valores de P H (a decir, RPH=−5mV/PH=−10mV y RPH=−10mV/PH=−20mV ) permiten deducir que el efecto sobre la corriente de fondo no supera el aumento teórico en la señal electroquı́mica por efecto de aumentar P H, lo que indica que al menos para estos valores de P H, el incremento en la señal electroquı́mica no se ve comprometida por el aumento en las corrientes de fondo. Resultados adicionales que apoyan esta observación se discutirán en la Sección 3.1.2. Una vez esclarecido el origen capacitivo de las corrientes de fondo en SWV, es posible obtener curvas maestras para realizar un procedimiento de sustracción sobre las señales resultantes de un proceso de transferencia. Esto no es del todo confiable en CV ya que debido a la falta de un ajuste adecuado de la corriente de fondo con respecto a la velocidad de barrido no se puede fincar el origen de éstas a un fenómeno 47 Tabla 3.1: Relación de corrientes de fondo en SWV a distintos P H n PH Pendiente, m mn−1 /mn Ψn Ψn−1 /Ψn 0 −5 mV 0.1009 - 0.1043 - 1 −10 mV 0.2164 0.4652 0.1984 0.5257 2 −20 mV 0.4352 0.4984 0.3744 0.5300 meramente capacitivo. La curva maestra que se utilizó durante la experimentación corresponde a la mostrada en la Figura 3.5. La discusión sobre los parámetros seleccionados para esta curva maestra se discuten en la sección 3.1.2. Figura 3.5: Curva maestra para la sustracción del fondo en SWV, T=26o C±1o C , P H=−20 mV, f =10 Hz. Barrido de 0.05 V a 0.85 V. La Figura 3.5 muestra adicionalmente la desviación estándar a partir de 4 mediciones independientes de la corriente de fondo, la cual se sitúa en un valor aceptable (± 0.1 µA) para la mayor parte del barrido. Algunas desviaciones mayores en la región comprendida entre 0.2 y 0.4 V tendrán un efecto sobre mediciones 48 subsecuentes (Sección 3.3.3). Esta determinación se tomó como base para las sustracciones de fondo; a menudo (y debido a valores dispares de la resistencia sin compensar que ocasionan distintas correcciones en el potencial) se observaba que en la experimentación se obtenı́an valores mayores de las corrientes de fondo en los extremos de la ventana de potencial (principalmente en aquellos mayores a 0.75 V). Las mediciones confiables entonces quedaron restringidas principalmente en los lı́mites comprendidos entre 0.15 V y 0.75 V, lo cual es un resultado acorde con lo observado en CV. 3.1.2. Obtención de parámetros óptimos Los parámetros utilizados en las técnicas como se muestran en las Secciones 2.2.2 y 2.2.3 se determinaron a partir de un análisis cualitativo y cuantitativo de la señal electroquı́mica. Cuantitativamente una comparación útil es la siguiente: en CV el valor para la corriente de fondo a ν= 20 mV/s es equiparable al valor de la corriente de fondo para f =10 Hz y P H=−20 mV en SWV, pero la ganancia en la señal electroquı́mica es notablemente diferente. Por ejemplo, para la transferencia de TMA+ 50µM , la intensidad de señal en CV para ν= 20 mV/S es de −3.20 µA (Sección 3.2), en SWV es de 3.42 µA para P H=−10 mV y se incrementa a 6.13 µA para P H=−20 mV. Ası́ pues, SWV ofrece la posibilidad de incrementar la sensibilidad del método mediante el ajuste de P H. Cualitativamente, se observó que la reproducibilidad de las mediciones se veı́a favorecida por el uso de un P H mayor en SWV. Una limitación instrumental en SWV fue el uso de un P H no mayor a −20 mV, ya que a excitaciones mayores la señal en las orillas de la ventana de potencial aumentaba significativamente y rebasaba el rango de corriente impuesto de 100 µA. Esto sucede ya que a pesar de que la corriente neta en SWV no sobrepasa los 20 µA, las componentes sı́ lo hacen, y causan una sobrecarga en el potenciostato que pone en marcha los sistemas de control del aparato que hacen que de medir la corriente. En el caso de CV, a menudo se observó que el aumentar la 49 velocidad de barrido no generaba una ganancia significativa en la señal, más aún, la reproducibilidad de la señal se veı́a afectada a velocidades mayores a 15 mV/s. Para estabilizar una señal a una velocidad de barrido mayor a 15 mV/s se debı́an correr al menos 3 ciclos, lo cual resulta en una desventaja al compararla con SWV en donde el barrido se realiza en 1/8 del tiempo requerido para los 3 ciclos de CV. A cualquier velocidad de barrido utilizada en CV y en general para concentraciones del analito menores a 100 µM la señal resultante presentó un achatamiento en la corriente de pico. Este achatamiento llega a abarcar hasta 10 puntos experimentales, mismos que a un incremento en el escalón de 2 mV por punto implican una incertidumbre de 20 mV en la determinación del pico. En SWV la corriente de pico no abarca más de 2 puntos experimentales, por lo que la determinación de este está sujeta a una menor incertidumbre. La mayor sensibilidad de la técnica de SWV se evidencı́a bajas concentraciones del analito, en las que se presenta una ganancia importante en términos de la señal electroquı́mica y de la facilidad de lectura del voltamperograma. Las Figuras 3.6 y 3.7 muestran una comparación entre las señales electroquı́micas en ambas técnicas para TMA+ = 12.5 µM . SWV presenta una curva caracterizable mientras que en CV el proceso no se puede observar incluso aumentando la velocidad de barrido. En resumen, para CV se prefirió el uso de ν=10 mV/s al no haber una ganancia significativa a mayores velocidades de barrido sin comprometer la reproducibilidad del resultado; en SWV se prefirió P H=−20 mV en términos de la ganancia de la señal y de la reproducibilidad. La frecuencia f = 10 Hz se fijó debido a la simetrı́a de la señal obtenida y para evitar un incremento mayor de las corrientes de fondo. Estos parámetros son similares a los reportados para otros estudios [18], [28]. 50 Figura 3.6: Sensibilidad en CV para analito a bajas concentraciones. Para TMA+ 12.5 µM la señal no es distinguible inclusive a velocidades de barrido mayores a ν= 10 mV/s. 3.2. Evaluación de la transferencia por CV La técnica de CV resultó útil para el análisis de la transferencia de iones a altas concentraciones (mayores a 100 µM ). A bajas concentraciones la determinación de la corriente de pico (y por lo tanto la determinación de los potenciales de pico para obtener el potencial de onda media) es difı́cil debido a un achatamiento de la señal. La Figura 3.8 muestra la transferencia de TMA+ evaluada por CV. Si bien a concentraciones mayores a 50 µM se observa un pico bien definido en la dirección de barrido hacia potenciales positivos, en el barrido reverso se presenta una transferencia en dos pasos. La separación entre las intensidades de pico principales (a decir, las mayores) es en general superior a 160 mV, lo cual no concuerda estrictamente con la descripción de un proceso reversible (Sección 1.2) sin embargo, a concentraciones más altas la aparición de un segundo pico más cercano en el eje de 51 Figura 3.7: Sensibilidad en SWV para analito a bajas concentraciones.Para TMA+ 12.5 µM la señal es distinguible inclusive a la frecuencia más baja f =10 Hz. potencial al pico en el barrido positivo concuerda bien con la separación esperada de 59 mV. En la literatura no se halla reportado el caso de la transferencia de TMA+ a bajas concentraciones a manera de comparar este resultado que a primera vista resulta anómalo; más aún, la propuesta de que un proceso de adsorción en la interfase que predomine sobre el proceso de difusión resulta sugerente aunque descartado por mediciones de tensión superficial mediante la técnica de Impedancia Electroquı́mica [29] al menos para concentraciones altas del ión de estudio. La separación de picos mayor a 59 mV sin embargo si está reportada en estudios de transferencia de otros iones a bajas concentraciones [18], [28]. Los resultados de la transferencia por CV (Figura 3.8) pueden ser analizados desde el punto de vista analı́tico a través de una linealización de Randles-Sevcik (Ecuación 1.6) con respecto a la concentración. Para dicho análisis se ha considerado solamente la intensidad de pico correspondiente al barrido en la dirección positiva, los resultados se muestran en la Figura 3.9. La Figura 3.9 muestra una una buena correspondencia con 52 Figura 3.8: Transferencia de TMA+ evaluado por CV, ν= 10 mV/s. el modelo lineal, sin embargo hay dos aspectos importantes a considerar. El primero es que se ha omitido la lectura a concentraciones menores a 25µM debido a que la determinación de la intensidad de pico es poco clara dado el achatamiento de la señal. El segundo aspecto es que la determinación del potencial de transferencia (el potencial de onda media en este caso) está sujeto a una gran incertidumbre. Por una parte, la observación del proceso de transferencia en dos pasos introduce la interrogante sobre qué pico se debe elegir para realizar la determinación del potencial de onda media, y por otra parte, inclusive en el barrido electo se observa un desplazamiento de la señal en el eje del potencial, lo cual introduce una incertidumbre aún mayor. La pérdida de información sobre el sistema nos previene entonces de obtener mediciones confiables a partir de la técnica de CV para los fines analı́ticos que persigue este estudio. Para la transferencia de CH+ se observó la misma complicación que para el TMA+ . Para la transferencia de TBA+ una complicación adicional surge al no observar el proceso de transferencia en la segunda parte del ciclo, lo cual imposibilita la determinación del potencial de onda media. La Figura 3.10 muestra los resultados para la 53 Figura 3.9: Linealización de Randles-Sevcik para la transferencia de TMA+ , ν= 10 mV/s. transferencia de éste catión evaluados por CV para una concentración relativamente alta (150µM ). La sensibilidad ofrecida por la téncia no permite una observación clara del proceso e transferencia inclusive para el ciclo de polarización hacia potenciales positivos. El obtener una medición confiable y más sensible es el principal reto que se plantea en la transferencia evaluada por SWV, Sección 3.3. 3.3. Evaluación de la transferencia por SWV Una introducción a la ventaja analı́tica que presenta la técnica de SWV ya ha sido dada en la Sección 3.1.2, en la que la determinación de los parámetros óptimos de análisis responde principalmente a la búsqueda de la reproducibilidad en los datos y en la mejora de la respuesta electroquı́mica. Los resultados mostrados en las secciones a continuación son producto de un proceso de mejora en las condiciones experimentales y del reconocimiento de los factores que afectan determinantemente a la reproducibilidad de los datos. En gran medida el reto que representó la mejora de 54 Figura 3.10: Transferencia de TBA+ 150µM evaluado por CV, ν= 10 mV/s. El pico encerrado en el cı́rculo corresponde a la intensidad máxima que se percibe, este resultado corresponde al tercer barrido. la señal electroquı́mica al utilizar la técnica de SWV se dio en el terreno del arreglo experimental como ya se ha mencionado en la Sección 2.1.4. Los parámetros utilizados para SWV corresponden a los descritos en la Tabla 2.3. 3.3.1. Transferencia de TMA+ La transferencia de TMA+ evaluada por SWV se muestra en la Figura 3.11 en la que se presentan las curvas promedio para 6 mediciones por cada concentración de analito; la señal de transferencia es bastante clara inclusive sin haber realizado la sustracción de fondo. La respuesta electroquı́mica sigue la tendencia predicha por la Ecuación 1.13 al incrementarse ∆Ip con respecto a la concentración del analito en la fase acuosa. 55 Figura 3.11: Transferencia de TMA+ evaluada por SWV sin sustracción del fondo. Ep = 0.560 V ± 0.015 V. A partir de una concentración mayor a 100µM se observa un cambio importante en el comportamiento de la curva en el sentido que no sigue el mismo patrón de crecimiento que las demás. A concentraciones mayores a 200 µM la técnica muestra una pérdida de la sensibilidad caracterizada por una disminución marcada de ∆Ip . Es en este punto precisamente cuando la técnica de CV resulta adecuada para realizar una medición; ası́ pues, ambas técnicas se complementan en disntintos rangos de concentración. La motivación del presente estudio, sin embargo, es lograr una sensibilidad adecuada a bajas concentraciones, por lo que es conveniente enfocarse en este aspecto. Cuando se realiza la sustracción del fondo (Figura 3.5) a las curvas de la Figura 3.11 se obtiene una secuencia de curvas corregidas como las mostradas en la Figura 3.12. Estas curvas corregidas presentan la ventaja de poder analizar su forma y determinar su calidad (Sección 3.4) después de eliminar la contribución de la corriente capacitiva. 56 Figura 3.12: Transferencia de TMA+ evaluada por SWV con sustracción del fondo. Ep = 0.560 V ± 0.015 V. A partir de las curvas corregidas que se muestran en la Figura 3.12 se pueden determinar por inspección los valores para ∆Ip y realizar la linealización correspondiente con respecto a la concentración como se muestra en la Figura 3.13. La Figura 3.13 muestra un buen acuerdo con el modelo lineal. Es importante hacer mención que el análisis presentado en algunas publicaciones [28] sugieren el uso de un ajuste a una función de logaritmo natural, mismo que en general no resulta tan bueno como el lineal aquı́ presentado. Otros ajustes, en el caso que la adsorción del ión en la interfase resultara importante, se intentaron. El ajuste al modelo de Langmuir falla, y en cuanto a la función logarı́tmica que representarı́a un modelo de adsorción de Frumkin, no resulta adecuado. En este sentido, es importante reconocer la diferencia entre la pérdida de sensibilidad de la técnica a concentraciones mayores a las presentadas en la Figura 3.13 de la necesidad de introducir una función de ajuste diferente. Resulta entonces apropiado añadir que la respuesta lineal sólamente es válida en el intervalo entre 12.5 y 200 µM . 57 Figura 3.13: Linealización de ∆Ip contra la concentración de TMA+ . Existe una buena correlación con el modelo lineal. Puede notarse que la desviación estándar, incluyendo la contribución del fondo, es prácticamente del orden del tamaño de los sı́mbolos. 3.3.2. Transferencia de CH+ La obtención de las curvas para CH+ se efectuó de una manera análoga al TMA+ . Una pequeña cantidad de TMA+ como referencia interna se adicionó a las muestras de CH+ con la intención de referenciar el potencial de transferencia de estas. Esto no se logró debido a que los potenciales de transferencia de ambos iones son similares (una discusión más profunda se presenta en la sección 3.4). La sustracción de fondo se hizo considerando la cantidad añadida de TMA+ ; en algunas muestras pequeñas cantidades del ion de referencia se observan aún, aunque en general no afectaron de manera apreciable los cálculos posteriores. Cada curva representa el promedio de seis mediciones. La Figura 3.14 muestra los resultados para esta transferencia. El potencial promedio de pico de 0.65 V que implica una separación de 0.09 V con respecto al potencial promedio de pico del TMA+ que se observó en la celda 58 corresponde bien a la diferencia reportada en la literatura, donde ∆φCH+ − ∆φTMA+ = 0.082 V [1]. Figura 3.14: Transferencia de CH+ evaluada por SWV con sustracción del fondo. Ep = 0.650 V ± 0.011 V. La linealización para ∆Ip con respecto a la concentración del analito se muestra en la Figura 3.15, de nuevo los puntos se ajustan bien al modelo lineal. Para este caso hubo un control más apropiado de las condiciones experimentales (por ejemplo, mejor control de la temperatura, reposo del sistema después de cada barrido y aislamiento del sistema introduciéndolo a la caja de Faraday). Un mayor cuidado en dichas condiciones mostró mejores resultados que en el caso del TMA+ con el cual se habı́a trabajado de manera extensa durante otras etapas de la investigación. 59 Figura 3.15: Linealización de ∆Ip contra la concentración de CH+ . Existe una buena correlación con el modelo lineal, las barras de error indican ± desviación estándar incluyendo la contribución del fondo. 3.3.3. Transferencia de TBA+ En el caso de la transferencia de TBA+ la obtención de resultados cuantitativos fue un reto que se superó con la sensibilidad de la técnica de SWV. La determinación cualitativa y cuantitativa de TBA+ se complica debido a que su señal se empalma con el extremo de la ventana de potencial que corresponde a la transferencia de cationes hidrofóbicos. De hecho, el TBA+ fue utilizado extensamente como catión de soporte orgánico antes del uso de otros cationes más hidrofóbicos (como el BTPPA+ en el caso de esta investigación). En la Sección 3.2 se mostraba que la señal en CV para la transferencia de TBA+ era débil y difı́cil de distinguir de la ventana de potencial, más aún, el potencial de media onda no se podı́a determinar debido a la falta de la onda reversa. La Figura 3.16 muestra la transferencia de TBA+ evaluado por SWV para tres concentraciones selectas. Cuando los resultados de la Figura 3.16 se someten a la sustracción del fondo (Sección 3.3.1), la transferencia del ión se 60 Figura 3.16: Transferencia de TBA+ evaluada por SWV sin sustracción del fondo. Si bien la señal se presenta empalmada con la ventana de potencial, a concentraciones menores a 100µM se logra observar el proceso. TMA+ añadido a manera de referencia. observa con claridad. La Figura 3.17 muestra la transferencia; el aumento de la señal electroquı́mica con respecto a un aumento de la concentración es evidente. El potencial promedio de pico de 0.27 V que implica una separación de −0.29 V con respecto al potencial promedio de pico del TMA+ que se observó en la celda corresponde bien a la diferencia reportada en la literatura, donde ∆φTBA+ −∆φTMA+ = −0.286 V [1]. La linealización de ∆Ip con respecto a la concentración se presenta en la Figura 3.18. El ajuste lineal es adecuado aunque no tan bueno como en el caso del CH+ y TMA+ . El cálculo de la desviación estándar para cada punto viene dado por la suma de la señal y el fondo; en la Figura 3.5 ya se anticipaba que esta región de la ventana de potencial estaba sujeta a una mayor variación que otras regiones. Resulta desafortunado que sea éste intervalo de potencial en el que aparezca la señal para el TBA+ . Es importante enfatizar, sin embargo, que la observación clara del proceso de transferencia después de la sustracción de fondo y el hecho de que las curvas 61 Figura 3.17: Transferencia de TBA+ evaluada por SWV con sustracción del fondo. La calidad de la señal se ve ampliamente favorecida por el uso de la técnica de pulsos. Ep = 0.270 V ± 0.010 V. TMA+ Ep = 0.560 V añadido a manera de referencia. resultantes se puedan caracterizar (Sección 3.4.1) es un logro importante en este estudio, sobre todo para las bajas concentraciones utilizadas del analito. 3.3.4. Análisis estadı́stico de las curvas de calibración Las curvas de calibración obtenidas en las secciones 3.3.1, 3.3.2 y 3.3.3 de acuerdo a su grado de ajuste influirán en la exactitud con la que se puede determinar una muestra desconocida. El grado de ajuste se ha evaluado a partir de los valores de R2 . Sin embargo, R2 solo indica estadı́sticamente hasta qué grado la linealización explica el error. Es importante que no se observe una tendencia que ponga en duda el modelo lineal. Esto se puede evaluar mediante el comportamiento de los residuales. Los residuales indican la diferencia entre un determinado punto en la curva de calibración y el valor predicho por la linealización. Una falta de tendencia en ellos indica que en 62 Figura 3.18: Linealización de ∆Ip contra la concentración de TBA+ . Existe una correlación adecuada con el modelo lineal, las barras de error indican ± desviación estándar incluyendo la contribución del fondo, en éste caso hay una variabilidad considerable en cada punto. efecto, las diferencias o residuales, se deben a la dispersión de datos producto de la incertidumbre. Las Figuras 3.19, 3.20 y 3.21 muestran los residuales para las curvas de calibración presentadas en las secciones precedentes. En general, no se observa ninguna tendencia de importancia en ellas [30]. Para la obtención de los lı́mites de detección (LDM) y de cuantificación (LCM) del método, se puede utilizar la información contenida en las curvas de calibración para determinar el valor de la desviación estándar en las mediciones de corriente. La necesidad de calcular la desviación estándar sigue a las fórmulas para obtener dichos lı́mites: LDM = yb + 3sy (3.4) LCM = yb + 10sy (3.5) Para estas dos ecuaciones, nos valdremos entonces la desviación estándar que se presenta en las curvas de calibración, y como valor base yb se utilizará el intercepto con 63 Figura 3.19: Gráfica de Residuales para la curva de calibración de TMA+ . No hay una tendencia clara en los Residuales. el eje de ∆Ip . La desviación estándar en el eje de ∆Ip se puede calcular mediante la siguiente fórmula [30]: sy = D − m2 C n−2 !1 2 (3.6) donde m es la pendiente de la gráfica y el término de n-2 se introduce al indicar que el cálculo consume dos grados de libertad (uno correspondiente a la pendiente y otro al intercepto) de el número de puntos n en la curva de calibración. D y C representan la diferencia de cuadrados en la ordenada y abscisa respectivamente, asignándole el término y a las ordenadas y x a las abscisas se escribe: D= X C= X ( 2 ( y − x − y)2 n (3.7) x)2 n (3.8) P 2 P La determinación de sy se llevó a cabo en una hoja de cálculo. La Tabla 3.2 resume los resultados para el análisis. La Tabla 3.2 muestra la repercusión de un ajuste lineal menos adecuado en el caso del TBA+ ya que el LDM y el LCM son sustancialmente mayores que para los otros 64 Figura 3.20: Gráfica de Residuales para la curva de calibración de CH+ . No hay una tendencia clara en los Residuales. Tabla 3.2: LDM y LCM calculados para las curvas de calibración. Sustancia sy /µA yb /µA LDM /µM LCM /µM TMA+ 0.2851 1.1765 11.31 37.50 CH+ 0.1549 0.6971 7.04 23.47 TBA+ 0.2229 0.8488 16.91 53.95 dos iones. Al comparar estos resultados con los obtenidos en CV en los que inclusive la transferencia para una concentración tan alta (comparada con las estudiadas en esta sección) de 150 µM es poco clara, se demuestra el mayor poder analı́tico de la técnica de SWV. Un aspecto importante a considerar en las ecuaciones de ajuste obtenidas es que el valor de los interceptos en general es mucho mayor que el valor de las pendientes. Esto es algo desconcertante en el sentido del procedimiento llevado a cabo, en el que la sustracción del fondo deberı́a minimizar dicho intercepto, sin embargo, una inspección rápida al procedimiento de análisis presentado en la Sección 3.1.1 respectivo a las 65 Figura 3.21: Gráfica de Residuales para la curva de calibración de TBA+ . Para concentraciones bajas del analito parece haber una tendencia que después es rota por valores a más altas concentraciones. La linealización no es tan adecuada como para las otras sales de estudio, sin embargo no presenta una tendencia clara. corrientes de fondo muestra que inclusive en ausencia de un analito existe una señal residual que no se elimina. En el caso de las corrientes de fondo, se deberı́a cumplir igualmente que a una frecuencia f o velocidad de barrido ν nula la señal resultante fuera mı́nima, sin embargo no es el caso. Los valores de R2 obtenidos para las curvas de calibración sugieren que gran parte del alto valor de los interceptos de debe a un ajuste que en general resultó adecuado al mostrar una tendencia clara, pero deficiente en términos estrictos. Los factores más importantes que se pueden encontrar como generadores de la variabilidad de los datos entre una celda y otra (e inclusive en repeticiones del análisis de una muestra en la misma celda) son numerosos. Un gran esfuerzo experimental se realizó para minimizar el efecto que éstos pudieran tener. Para una discusión más a fondo debemos recurrir a la Ecuación 1.13 que predice el valor para la corriente diferencial de pico. La Ecuación 1.13 incluye un término de Área, el cual se puede considerar como un área efectiva de transferencia que será proporcional al área ge- 66 ométrica. El uso de un área pequeña para la interfase, por ejemplo, al comparar la celda grande con la celda pequeña cuyo radio de áreas es de 2.4, obedece a la suposición de que el control del potencial a través de la interfase es mejor al tener una discrepancia menor entre los capilares de Luggin y la interfase. Esta relación, sin embargo, debe ser suficiente para obtener una señal adecuada. Para obtener resultados reproducibles se debe asegurar que el área que ocupa la interfase sea constante entre experimentos, sin embargo, no se encontró otro método más que la inspección visual para determinar la planaridad de la interfase. De hecho, esta podı́a ser modificada a voluntad mediante la remonición de pequeñas cantidades de las disoluciones de referencia orgánica o de trabajo. Ahora bien, la planaridad de la interfase no sólo garantiza que entre celdas distintas se tenga el mismo factor geométrico, sino que también permite la obtención de resultados similares para la compensación IR. La compensación IR en este sentido es una técnica que si bien libra al experimento de un error mayor, a su vez puede introducir un factor difı́cil de controlar. Variaciones sustanciales en la compensación IR ocurren si el arreglo de electrodos es muy diferente, pero inclusive con variaciones de entre 100 y 200 Ω se pueden generar errores significativos, sobre todo para la transferencia a concentraciones intermedias y altas. Dos valores diferentes para la compensación IR generarán una diferente medición en el potencial y por lo tanto, la corriente que se mide en cada paso de potencial, dando lugar a variaciones en la forma de la gráfica y eventualmente a la posición y valor de la corriente de pico en SWV o en CV. Lo mismo es válido al hablar de la calidad de los electrodos de referencia, cuya función es en sı́ el regular el potencial a través de la interfase: si estos no son lo suficientemente buenos pueden introducir un error aún mayor que la compensación IR. Un gran avance se detalla en la sección 2.1.4 en cuanto al mecanismo de llenado de la celda; dicho sea que uno de los retos más grandes que se enfrentaron en el periodo experimental fue precisamente la obtención de datos reproducibles, lo cual se logró en gran medida. Un factor que resulta de gran importancia en esta discusión es el rol que juega el control de la temperatura, mismo que se discutirá con mayor profundidad en la Sección 3.5. El control de temperatura de ± 1o C y a veces de ± 2o C dependiendo de las condiciones atmosféricas resulta 67 ser un factor definitivo, cuyo impacto se subestimó hasta el estudio controlado de la transferencia a distintas temperaturas. Un control adecuado de temperatura debe establecerse con una precisión de ±0.1 o C. A pesar de las inconsistencias mencionadas, las calibraciones son funcionales cuando se llevan al terreno experimental. Esto indica que, a pesar de que el análisis matemático no es del todo satisfactorio, en la práctica la aplicación analı́tica sı́ tiene un valor de uso. La aplicación de las curvas se puede ilustrar bien a través de un ejemplo. Ejemplo de aplicación para la cuantificación de una muestra de TMA+ 50 µM Se llevó a cabo la cuantificación de una muestra de TMA+ en dos celdas a 22o C, se tomaron 4 mediciones por cada celda. Los resultados para las corrientes resultantes después de la sustracción del fondo y de una correción para la temperatura (Sección 3.5.2) para fijarla referenciarla a 25o C se muestran en la Tabla 3.3. Tabla 3.3: Análisis de [TMA+ ]= 50µM con corrección. Determinación ∆Ip,corr / µA 1 4.82 2 4.74 3 4.70 4 4.68 5 4.90 6 4.83 7 4.85 8 4.91 Para el análisis de los datos, calculamos la desviación estándar y el promedio tanto para ∆Ip como para su transformación en la concentración. Enseguida, calculamos el 68 intervalo de confianza (LC) que se define de la siguiente manera: LC = ∆Ip ± st(0.95,n−1) n1/2 (3.9) El valor de n es 8 (son 8 determinaciones) y el valor para la función t de Student para un nivel de significancia de 95 % para 7 grados de libertad es de 2.365. Un nivel de significancia de 95 % implica que si hiciéramos 20 juegos de determinaciones diferentes, esperarı́amos que 19 de ellos se situarı́an en el intervalo fijado por la media poblacional y el lı́mite de confianza. Los resultados del análisis se muestran en la Tabla 3.4. Tabla 3.4: Análisis estadı́stico para [TMA+ ]= 50µM Parámetro Valor ∆Ip 4.80 µA s∆Ip 0.0876 µA [TMA]+ prom 47.42 µM sTMA+ 1.15 µM Lim Confianza ± 0.96 µM LDM 3.43 µM LCM 11.45 µM El análisis muestra que el método es funcional, se tiene una recuperación de 94.82 % y un lı́mite de confianza de aproximadamente ± 1 µM . Ahora bien, en la Tabla 3.4 se nota el uso de un LDM y un LCM adicionales; estos provienen de la desviación estándar en las mediciones hechas y no del error introducido por la curva de calibración que se ve reflejado mejor en el valor de la recuperación. Esta determinación fue hecha en la última etapa de la experimentación, una vez que una pieza clave de información con respecto a la temperatura se contempló (Sección 3.5.2) y se muestra mucho mejor que los parámetros calculados con anterioridad; a pesar de que estos datos son válidos, no es apropiado ignorar el error introducido 69 por la curva de calibración y reflejado en los mayores valores para LDM y LCM , por lo que estos se toman para propósitos prácticos como el valor reportado por este estudio. Vale la pena señalar, sin embargo, que el control introducido a través de un baño de temperatura constante (que bien no tiene la precisión de ± 0.1 o C deseado) y el poder referenciar las mediciones a la temperatura de trabajo de la curva de calibración mejoran notablemente el desempeño analı́tico de la técnica. 3.4. Evaluación de la calidad de las curvas en SWV y empalmes En las secciones precedentes se ha hecho un análisis de la respuesta electroquı́mica basado en el comportamiento de ∆Ip sin embargo es posible analizar también la forma de las curvas para obtener mayor información sobre el sistema. En las Sección 3.4.1 se comprobará la calidad de las curvas obtenidas para la transferencia de un sólo ión de estudio mientras que en la Sección 3.4.2 se analizará el caso en el que dos señales se empalman. El método de análisis que se tratará consiste en una linealización que permite evaluar la calidad de la gráfica resultante, en general buenos ajustes resultan para la transferencia de un ion, mientras que los empalmes causan desviaciones de la linealidad. 3.4.1. Análisis de curvas en SWV El análisis de las curvas como es propuesto por Aoki et al. [31] sugiere la descomposición de la resolución general para la respuesta electroquı́mica en SWV en sus componentes de muestreo de corriente y geométrica. La corriente diferencial ∆I en estos términos está descrita por la componente de muestreo de corriente H(A, m, τ ) y su componente geométrica G(ζ) de la siguiente manera: ∆I = nF C ∗ H(A, m, τ )G(ζ) 70 (3.10) donde las funciones H(A, m, τ ) y G(ζ) están definidas de la siguiente manera: ( H(A, m, τ ) = A − 2 2m X ! ) j (−1) f ((j/2)τ ) + f ((m + 1/2)τ ) (3.11) j=1 !−1 G(ζ) = 1 + exp(−ζ − ζSW ) !−1 − 1 + exp(−ζ + ζSW ) (3.12) A es el área del electrodo, m es un entero que depende del muestreo de la corriente medida, j es un término de recursividad, τ es el inverso de la frecuencia f y los términos de ζ se definen de la siguiente manera: nF ESW RT ζSW = (3.13) 0 nF (E − E ◦ ) ζ= RT (3.14) 0 E es el potencial de medición en tanto E ◦ es el potencial formal (a decir, el potencial de pico Ep ). En el caso del análisis de la forma de la gráfica, nos interesa la componente G(ζ). Se puede notar que el término de concentración, C ∗ y de frecuencia 1 τ son independientes de la esta función, por lo que el análisis se puede aplicar a todas las curvas obtenidas. Ahora bien, si la Ecuación 3.10 se resuelve con respecto al potencial eliminando la dependencia de H(A, m, τ ) (misma que indica la intensidad de la respuesta) se obtiene la siguiente ecuación: 2.3RT E=E + nF ◦0 ! 2 log z − (z − 1) 1 2 ! (3.15) Se define a la función z como: z= ∆Ip ∆I ! ! 1 + cosh ζsw − cosh ζsw (3.16) Nótese que la función z referencı́a cualquier valor de ∆I al valor de ∆Ip lo cual es una manera de reducir este valor. Para un pulso de potencial de −20mV (ESW = −20 mV), una temperatura de 298 K y en nuestro caso la transferencia de un catión monovalente, ζSW = 0.7780. 71 La linealización propuesta por la Ecuación 3.15 se aplicó a las curvas sustraidas obtenidas por SWV para la mitad de su recorrido (observando que tienen una simetrı́a adecuada). Idealmente, se espera que el intercepto muestre el potencial de pico (Ep ) mientras que la pendiente tenga un valor de 59 mV. Para los valores que nos interesan de ∆I se eligió como primer punto el principio de la curva y como punto final uno antes del potencial de pico. En el eje de las ordenadas se eligió colocar al Potencial E y en el eje de las abscisas la función de z. El tratamiento numérico se hizo en una hoja de cálculo y las linealizaciones se analizaron mediante el método de mı́nimos cuadrados eligiendo el valor de R2 como indicador de la bondad del ajuste. El análisis para la transferencia de sólamente un ion se presenta en las Figuras 3.22, 3.23 y 3.24 a través de un ejemplo para cada uno de los iones estudiados. Las Tablas 3.5, 3.6 y 3.7 muestran los resulados de las linealizaciones. Figura 3.22: Linealización para evaluar la forma de la curva, TMA+ 100µM . Las Figuras 3.22, 3.23 y 3.24 evidencı́an que el ajuste es adecuado, si bien no del todo ideal, como para la transferencia de CH+ , aunque es preciso mencionar que para este caso las curvas son el producto de la sustracción de una curva de TMA+ . En 72 Figura 3.23: Linealización para evaluar la forma de la curva, CH+ 100µM . general, las tablas 3.5, 3.6 y 3.7 muestran un buen acuerdo con el modelo lineal para el comportamiento de E contra f (z). Los interceptos en general convergen hacia el potencial de transferencia (Sección 3.3). En el caso de la pendiente, se observa una dispersión importante entre diferentes curvas. Para la mayorı́a de estas, el valor de la pendiente es superior al esperado de 59 mV, con excepción en el caso del TBA+ a bajas concentraciones. Para altas concentraciones de cualquier analito, se observa un aumento en el valor de la pendiente, mismo que es el reflejo del ensanchamiento de las curvas. El caso ideal que se propone a través de las Ecuaciones 3.10 a 3.16 implica que el coeficiente de difusión, en un sistema acuoso convencional, de la especie oxidada y reducida sea el mismo. En el caso de estudio de este trabajo eso no es necesariamente cierto, sobre todo en el sentido de que los equivalentes a especie oxidada y reducida son el ion en la fase orgánica y en la fase acuosa respectivamente. Se puede esperar que el diferente comportamiento del ion, implı́cito en los diferentes valores del coeficiente de difusión en una y otra fase introduzcan un error en estas consideraciones. La transferencia iónica, sin embargo, está ampliamente reportada 73 Figura 3.24: Linealización para evaluar la forma de la curva, TBA+ 100µM . como un fenómeno reversible (Sección 1.2), ası́ pues, el uso de las herramientas teóricas (como el caso de la presente linealización) en la técnica de SWV es válido, aunque debe tomarse con cautela al hacer consideraciones como las anteriores acerca del coeficiente de difusión. El uso de la linealización en esta sección está respaldada por el hecho de que es aplicable en sistemas similares a las ITIES, como el electrodo colgante de mercurio, y en general, a interfases planas y grandes en las que se puede presentar la solubilidad de la especie oxidada en el electrodo [31] (lo cual es una analogı́a útil en el presente trabajo). Los valores adecuados de R2 aquı́ descritos contrastan con los resultados para una transferencia mixta, como se evaluará en la siguiente sección. 3.4.2. Análisis de transferencias mixtas Cuando dos especies se transfieren a potenciales cercanos, se puede esperar que la señal electroquı́mica resultante sea la suma de los dos procesos, sin embargo, 74 Tabla 3.5: Indicadores de la calidad de las curvas de TMA+ [TMA]+ / µM R2 Pendiente / V Intercepto / V 25 0.9988 0.0875 0.547 50 0.9930 0.0939 0.541 75 0.9948 0.0878 0.537 100 0.9999 0.0957 0.550 140 0.9947 0.1327 0.541 Tabla 3.6: Indicadores de la calidad de las curvas de CH+ [CH]+ / µM R2 Pendiente / V Intercepto / V 10 0.9964 0.0748 0.658 20 0.9977 0.0781 0.652 60 0.9959 0.0870 0.652 80 0.9996 0.0840 0.650 100 0.9920 0.1092 0.656 estarán superpuestos. Esta superposición no permitirá una lectura fidedigna de los parámetros de pico requeridos para la identificación de dichas sustancias, más aún, si no se llevara a cabo un análisis de la señal electroquı́mica se podrı́a juzgar erróneamente la transferencia de una sóla especie siendo que hay más especies que se transfieren. Las Figuras 3.25 y 3.26 muestran simulaciones para cuatro casos tipo: cuando la diferencia de potenciales de transferencia no permite diferenciar dos señales de pico y cuando sı́ es posible. Las simulaciones se llevaron a cabo con dos señales de TMA+ de intensidad de 6µA colocando ∆Ip en diferentes posiciones para una de las curvas con respecto a la otra. E2 indica la señal móvil y E1 la fija. 75 Tabla 3.7: Indicadores de la calidad de las curvas de TBA+ [TBA]+ / µM R2 Pendiente / V Intercepto / V 40 0.9804 0.0551 0.274 60 0.9985 0.0575 0.274 80 0.9908 0.0807 0.263 100 0.9986 0.0760 0.277 150 0.9990 0.1248 0.291 La simulación en la Figura 3.25 muestra que a una diferencia de señales de pico mayor o igual a 0.1 V se presenta una deformación considerable en la curva a juzgar por el ancho de la base y la región cercana a la señal aparente de pico. Sin embargo, para diferencias menores entre las señales de pico, la forma de la gráfica no permitirı́a decidir por simple inspección visual si esta es correcta o no. Por su parte, en la Figura 3.26 se observa claramente que a una separación de al menos 0.2 V las intensidades de pico no se afectan considerablemente, es decir, existe una buena resolución para las curvas. Para decidir sobre la calidad de las señales en un proceso de transferencia mixta recurrimos de la misma manera que se presentó en la Sección 3.4.1 a una linealización del potencial contra una función de los parámetros de SWV. El procedimiento seguido fue el mismo, las mediciones se hicieron sobre la mitad izquierda de la curva en las simulaciones; cuando las señales de pico eran distinguibles se tomó la mitad izquierda de la curva recorrida y la intensidad del pico sobre esta señal con la finalidad de evaluar el efecto que tiene el decaimiento de la señal fija sobre esta. Una gráfica modelo se muestra en la Figura 3.27 para una separación de picos de 0.1 V (electa pues es similar al caso del empalme de TMA+ con CH+ ). La Figura 3.27 muestra que el ajuste lineal es muy deficiente. Es de especial interés notar que la linealización para la figura 3.27 es muy diferente a la Figura 3.23 76 Figura 3.25: Simulación de transferencia mixta, E2 − E1 ≤0.1 V. Nótese la distorsión de la señal electroquı́mica a medida que los potenciales de pico son más lejanos. En el caso de menor distancia entre potenciales de pico, la determinación de un proceso mixto serı́a difı́cil dada la forma aparentemente normal de la gráfica. en donde la falta de ajuste (aunque aún es adecuado) muestra desviaciones en la forma de la gráfica debido a otros factores experimentales. En la Figura 3.27 también es evidente la presencia de tres regiones en la gráfica: a bajos valores del potencial predomina la contribución de la curva desplazada, luego una región intermedia y finalmente a valores altos del potencial existe una contribución similar de la curva desplazada y la curva fija. Estas regiones se pueden identificar fácilmente teniendo como referencia la curva de linealización: la primer región se halla por encima de ella, luego la segunda se encuentra por debajo y para la tercera sube de nuevo. El comportamiento de esta linealización es constante para las simulaciones realizadas; en general los ajustes lineales son muy deficientes, lo que indica que este es un método 77 Figura 3.26: Simulación de transferencia mixta, E2 −E1 ≥0.15 V. Cuando la diferencia entre la señal de pico es de 0.2 V la intensidad de pico no se ve afectada por el otro proceso significativamente. sensible para determinar las transferencias mixtas. La Tabla 3.8 resume los valores de R2 para las simulaciones; nótese que a una diferencia de 0.2 V el valor de R2 aumenta y es similar a los presentados para las transferencias de un solo ion (Sección 3.4.1), lo cual concuerda bien con el grado de resolución que se obtiene para las señales de pico. El análisis de los datos experimentales para la transferencia mixta de CH+ y TMA+ concuerda bien con las observaciones hechas para las simulaciones. La Figura 3.28 muestra las curvas resultantes del proceso de transferencia mixta para estas dos especies. En la misma, se observa que a diferentes contribuciones de la concentración de CH+ la señal aparente de pico se desplaza más hacia el potencial de transferencia 78 Figura 3.27: Linealización para evaluar la calidad de una curva de transferencia mixta, simulación. Diferencia de 0.1 V en los potenciales de transferencia y misma contribución de cada una de las señales a la señal mixta. observado para la CH+ . Cuando la contribución de ambos iones es similar se observa mayor simetrı́a para la curva, sin embargo el ensanchamiento es apreciable al comparar con las transferencias de la sección 3.2. Al llevar a cabo la linealización para determinar la calidad de la curva, se observa una correspondencia con las tendencias mostradas en la Tabla 3.8 para las simulaciones. Más aún, las linealizaciones en sı́ muestran el mismo comportamiento que la Figura 3.27. Como comparación, podemos elegir el análisis de la curva de transferencia mixta para CH+ y TMA+ cuando tienen contribuciones similares; esto se muestra en la Figura 3.29. La similitud entre las Figuras 3.29 y 3.27 es evidente ya que en el caso experimental observamos las tres regiones alrededor de la curva de ajuste. El análisis llevado a cabo sin embargo, no nos aporta información sobre cual pudiera ser la identidad de las sustancias participantes, sólo nos indicará si hay una transferencia 79 Tabla 3.8: R2 para la evaluación de simulaciones de empalme. E2 − E1 /V R2 −0.02 0.9630 −0.05 0.9141 −0.10 0.9691 −0.15 0.9473 −0.20 0.9926 mixta. Si bien puede resultar un poco arbitrario asignar un valor de R2 ≥0.99 como indicación de un proceso de transferencia de un solo ion contra una transferencia mixta, este valor se mostró como uno que indica la diferencia entre un ajuste adecuado para los datos experimentales y un mal ajuste ya sea en las simulaciones o en las transferencias mixtas experimentales. Finalmente, la Tabla 3.9 muestra los valores de R2 para las curvas de transferencia mixta de la Figura 3.28. Tabla 3.9: R2 para la evaluación de la transferencia mixta de CH+ y 25µM TMA+ . [CH]+ / µM R2 10 0.9067 20 0.8653 60 0.8676 80 0.8876 100 0.9049 80 Figura 3.28: Transferencia mixta de CH+ y TMA+ manteniendo la concentración de TMA+ constante, E2 − E1 = 0.09 V. 3.5. Transferencia de iones a distintas temperaturas La última parte del presente estudio se enfocó en el comportamiento de la transferencia de iones a distintas temperaturas. La motivación principal de esta sección fue la idea de que a diferentes temperaturas se podrı́a separar dos señales en virtud de la diferente entropı́a de transferencia del ion; si bien para el arreglo experimental utilizado esto no fue cierto dado que se tiene que considerar que el potencial en la celda es el resultado de varios potenciales acoplados (al menos uno para cada interfase, Ecuación 2.1), se encontraron relaciones importantes tanto para la posición como la intensidad del pico en SWV que determinarán fuertemente la capacidad analı́tica de la técnica y que por otra parte explican parte del error en el ajuste de datos para la construcción de las curvas de calibración (Sección 3.2). 81 Figura 3.29: Linealización para la curva de transferencia mixta de CH+ y TMA+ . Concentración de TMA+ = 25µM y CH+ = 20µM , E2 − E1 = 0.09 V. Para el trabajo realizado en este apartado se hicieron modificaciones sustanciales al arreglo experimental. Se incorporó un sistema de recirculación y control de temperatura en el que se sumergió la celda electroquı́mica en agua/glicerina para mantener la temperatura constante. Debido a complicaciones instrumentales, principalmente debidas a la interferencia de los dispositivos electrónicos de un termómetro digital y del aparato de recirculación con los del potenciostato se tuvo que optar por una medición de temperatura con un termómetro de mercurio con una precisión de ± 1o C. A pesar de este control de temperatura, las mediciones son bastante confiables ya que gracias al recubrimiento que se hizo sobre el contenedor para la celda no se observaba un cambio de más de 1o C en la temperatura en un intervalo de 3 a 5 minutos después de estabilizar el sistema y la medición en SWV no toma más de 1 minuto. Fue también por esta razón que se hizo uso únicamente de la técnica de SWV para este apartado. Más detalles sobre el arreglo experimental se presentan en la Sección 2.1.5. 82 3.5.1. Comportamiento de las Corrientes de Fondo Las corrientes de fondo en la región de la ventana de potencial no aumentan significativamente con un aumento en la temperatura, sin embargo, en las orillas de la ventana sı́ hay un incremento apreciable que causa que la ventana se haga más estrecha a temperaturas altas. Este comportamiento se muestra en la Figura 3.30. Figura 3.30: Comportamiento de las corrientes de fondo a distintas temperaturas. La escasa variación de la corriente capacitiva (la que está propiamente comprendida en la ventana de potencial, es decir entre 0.2 y 0.7 V) indica que la capacitancia de la interfase no cambia drásticamente al variar este parámetro. Sin embargo, en las orillas de la ventana sı́ hay un cambio significativo. Las orillas de la ventana se forman debido a la transferencia de las sales de soporte, que es en sı́ una transferencia de iones, razón por la cual esperarı́amos que al aumentar la temperatura aumentara la intensidad de señal (por ejemplo, debido a un aumento en el coeficiente de difusión de las especies). Un aumento en la temperatura genera un ensanchamiento de la señal, razón por la cual a temperaturas más altas la ventana de potencial se hace más estrecha. 83 Una discusión más profunda sobre el comportamiento de la señal con respecto a la temperatura se encuentra en la Sección 3.5.2. Las curvas para las lı́neas base obtenidas a cada temperatura se utilizaron para realizar la sustracción del fondo a las mismas temperaturas medidas para los experimentos de tansferencia de iones. 3.5.2. Impacto de la temperatura sobre Ep En el caso de la transferencia de TMA+ y TBA+ a distintas temperaturas se observó un cambio sustancial en los Voltamperogramas resultantes. La tendencia más evidente es que hay un incremento en la señal correspondiente para ∆Ip y un desplazamiento en Ep . Una vez realizada la sustracción del fondo a cada temperatura con las mediciones hechas en la Sección 3.5.1, se obtiene la secuencia de Voltamperogramas mostrados en la Figura 3.31. Figura 3.31: Transferencia de TMA+ y TBA+ a distintas temperaturas. La señal de la derecha corresponde al TMA+ . 84 Figura 3.32: Linealización de los desplazamientos en el potencial para la transferencia de TMA+ . El desplazamiento del potencial que se observa corresponde al del potencial de la celda, no al potencial de transferencia de un ion. Esto es importante puesto que un desplazamiento en el potencial de transferencia implicarı́a que la pendiente de la gráfica de Potencial contra Temperatura multiplicada por el término zi F representa la entropı́a de transferencia del ión. Si los valores fueran obtenidos de tal manera resultarı́an de 3 a 4 veces mayores a los reportados mediante arreglos experimentales diferentes (no basados en una celda de cuatro electrodos) para interfases acuoso/orgánico y acuoso/orgánico-acuoso [32], [33]. La variación en los valores reportados en la literatura, sin embargo, ponen en entredicho la validez de cualquier técnica, incluı́da la del presente estudio; por ejemplo, para la entropia de transferencia del TMA+ se reportan valores entre 27.4 y 71.7 J/K · mol [33]. En realidad el desplazamiento que se observa en las Figuras 3.32 y 3.33 al modificar la temperatura de la celda es el producto de diversos procesos acoplados en donde el número mı́nimo de ellos será al menos igual al número de interfases involucradas en la medición del potencial presentes en el sistema. Ası́ pues, el comportamiento de cada uno de los electrodos de Ag/AgCl, 85 Figura 3.33: Linealización de los desplazamientos en el potencial para la transferencia de TBA+ . el comportamiento de la interfase del electrodo reversible de BTPPA+ , de la interfase principal y de las juntas lı́quidas contribuyen. Más aún, los coeficientes de actividad pueden cambiar de manera apreciable a distintas temperaturas contribuyendo a introducir una mayor variación en el valor de Ep , mismo que indica el potencial formal de transferencia. La tendencia lineal se entiende de la relación: ∆E = 1 (T ∆S − ∆H) zi F (3.17) Un procedimiento alternativo que se puede utilizar para eliminar el efecto de los potenciales acoplados es considerar lo siguiente: en una misma celda, como se hizo en este estudio, se puede obtener la diferencia entre las entropı́as calculadas a partir de las pendientes para las gráficas para uno y otro ion. Este procedimiento eliminarı́a los potenciales acoplados y nos dejarı́a con una diferencia de entropı́as, a decir: ∆tr STMA+ −TBA+ = ∆tr STMA+ − ∆tr STBA+ (3.18) Al realizar este cálculo, obtenemos que para el sistema estudiado a partir de las pendientes de las Figuras 3.32 y 3.33 multiplicadas por el término zi F , ∆tr STMA+ −TBA+ = 86 135 J/K · mol. A partir de datos reportados para las entropı́as de estos dos iones [33], ∆tr STMA+ −TBA+ = 155 J/K · mol a través de una medición con una celda a estilo de termopar acuoso/orgánico/acuoso y ∆tr STMA+ −TBA+ = 120 J/K · mol para el caso de la evaluación por CV. El resultado obtenido es intermedio a estos dos, ası́ que de cierta manera está en acuerdo con ellos. Un punto frágil en este cálculo es por supuesto la validez de las linealizaciones hechas dado que sus valores de correlación no son del todo buenos. A pesar de que no se puedan separar dos procesos diferentes de transferencia que aparecen empalmados a temperatura ambiente en virtud del dominio de la variación de otros potenciales de la celda sobre el de transferencia, el conocer la variación del potencial de pico es importante. Dicha importancia reside en el hecho de que el reconocimiento del ion que se analiza depende del potencial al cual se transfiera: un control inadecuado de la temperatura causará un error importante en la medición, este puede llegar a ser de hasta 10 mV en un intervalo de 5o C. Para la obtención de las curvas de calibración, se trabajó en un intervalo de aproximadamente 4o C, consecuentemente es posible que una parte sustancial de las desviaciones promedio observadas para el potencial se deban a esta dependencia de la temperatura. Existen, sin embargo, otros factores que afectan de igual o en mayor manera a esta determinación, por ejemplo, se observó que no dejar reposar la celda después de cada medición hasta su regreso a Voc llegaba a causar un error de hasta 15 mV en mediciones consecutivas. Finalmente, la menor bondad en el ajuste para el comportamiento térmico de la transferencia de TBA+ es producto en gran parte del descontrol presente para las corrientes de fondo alrededor de su potencial de transferencia, lo cual introduce acaso un error sustancialmente menor para el caso del TMA+ . 3.5.3. Impacto de la temperatura sobre ∆Ip Un segundo aspecto a considerar en el análisis de la transferencia a distintas temperaturas, y probablemente más importante debido al impacto que tuvo sobre 87 los resultados de la Sección 3.2 es la variación de ∆Ip . A pesar de carecer de un sustento teórico sólido para el ajuste de estos datos a un modelo lineal, los resultados se ajustan muy bien a este. Las linealizaciones correspondientes se muestran en la Figuras 3.34 y 3.35. Figura 3.34: Linealización de la variación en ∆Ip para la transferencia de TMA+ . La búsqueda de los factores que afectan ∆Ip en SWV no es una tarea sencilla como lo muestra el análisis a continuación. Una forma expandida para la predicción de ∆Ip es la siguiente [31] (en comparación con la Ecuación 1.13): 1 1 ∆ip = 0.9653zi F ACi∗ f 2 Di2 tanh(ζsw /2) (3.19) Donde todas las varibales tienen el significado utilizado a lo largo de este trabajo y ζsw ha sido definida en la Ecuación 3.13. Los valores de la función hiperbólica tanh(ζsw /2) pueden ser calculados para un P H= 20 mV y para el intervalo de temperaturas de estudio. La predicción del comportamiento de esta función de hecho indica una reducción en el valor de ∆Ip al aumentar la temperatura ya que en el intervalo analizado, va de 0.39 a 283 K a 0.32 a 353 K. El comportamiento de esta función entonces no 88 Figura 3.35: Linealización de la variación en ∆Ip para la transferencia de TBA+ . puede explicar el aumento en la señal para ∆Ip . Experimentalmente se observó que al aumentar la temperatura aproximadamente 20o C la forma de la interfase cambiaba ligeramente, curvéandose por efecto de la expansión del nitrobenceno. Este error se corrigió tomando pequeñas cantidades de la solución de referencia orgánica causando el aplanamiento de la interfase para evitar un aumento de la señal por un cambio en el área interfacial. El único factor que permanece y puede variar con la temperatura es el Coeficiente de Difusión del ión Di . Siguiendo la relación de Einstein se tiene que: 1 2 D = υRT zi F !1 2 (3.20) υ representa la movilidad iónica. La primer dependencia de la temperatura se observa con el término lineal dentro de los paréntesis de la esta ecuación. Sin embargo υ también es dependiente de la temperatura de una manera indirecta: υ= zi e 6πηa 1 ∝ T n e−Ea/RT η 89 (3.21) (3.22) η representa la viscosidad del medio, y se introduce a partir de la ecuación de StokesEinstein (una forma de la Ecuación 3.21 expresada directamente en términos de Di ). Como se muestra en la Ecuación 3.22, la viscosidad del medio depende de un término exponencial caracterizado por una energı́a de activación, por lo tanto decimos que la variación en la viscosidad del medio es un proceso activado. La introducción de un término preexponencial en la Ecuarión 3.22 nos provee una manera de explicar el comportamiento de los resultados experimentales, en tanto que el coeficiente de difusión sea proporcional a T 2 en una aproximación muy simplista. Una manera de expresar la dependencia de Di de la temperatura se puede resumir de la siguiente manera: D ∝ T m eEa/RT (3.23) Esta expresión reconoce simplemente que la difusión del ion es un proceso activado y que depende a su vez de un término de temperatura a un exponente definido; experimentalmente se obtiene que esta dependencia puede ser modelada entonces para el intervalo estudiado como una dependencia cuadrática (la raı́z cuadrada será un término lineal entonces). Esto resulta bastante conveniente ya que provee una aproximación sencilla a la referencia de las mediciones a una temperatura definida, por ejemplo, 25o C. La pendiente de las linealizaciones en las Figuras 3.34 y 3.35 explica en gran parte la variación en las mediciones usadas para la construcción de las curvas de calibración. En un intervalo de 4o C implica un error de ± 0.2µA, lo cual representa a bajas concentraciones hasta un 10 % de la señal observada en SWV, introduciendo ası́ un error considerable que puede ser bien el origen de un valor alto para el intercepto y también representar una buena parte del error en el ajuste lineal. A pesar de esto, el conocimiento de la dependencia de la temperatura nos permite corregir o referenciar mediciones hechas a distintas temperaturas y ası́ usar las curvas de calibración construı́das. El ejemplo mostrado en la Sección 3.3.4 hace uso de esta correción (las mediciones se tomaron entre 22o C y 23o C) y los resultados son adecuados. La aplicación de este resultado es importante en el sentido analı́tico de este trabajo ya que permitirı́a en principio la modificación de la temperatura de una celda para mejorar algunos procesos. Por ejemplo, la Figura 3.30 muestra que a bajas 90 temperaturas hay un achatamiento de las corrientes de fondo, misma que puede ser explotada para revelar con mayor facilidad un proceso de transferencia empalmado con los lı́mites de la ventana de potencial. En efecto, esto se observa con la transferencia de TBA+ evaluado en la sección anterior. La señal electroquı́mica también puede ser mejorada en el sentido de aumentar ∆Ip a costa de la temperatura (sin necesidad de recurrir a un P H mayor que cause problemas instrumentales), ya que el aumentar 9o C la temperatura de medición causará una ganancia de 1 µA en la misma. Desde el punto de vista teórico, la dependencia de la señal pone de relieve algunas caracterı́sticas sobre la transferencia iónica a las que tal vez no se les ha puesto demasiada atención, como el efecto del cambio en la viscosidad de las fases sobre la interfase o inclusive el cambio en las propiedades de los iones en un ambiente diferente, por ejemplo, la solvatación de estos (desde la perspectiva de υ). 91 Capı́tulo 4 Conclusiones El análisis cuantitativo y cualitativo de iones de amonio cuaternario es posible mediante la técnica de transferencia iónica a través de la interfase de dos electrolitos lı́quidos, aunque bien está sujeta a diversas complicaciones experimentales que imponen restricciones a la precisión del análisis. Entre las complicaciones analı́ticas más importantes destacan la persistencia de corrientes de fondo en las mediciones, la variabilidad de los resultados entre diferentes celdas, la alta calidad de los instrumentos y procedimientos necesarios para llevar a cabo la experimentación y el alto impacto de la variación de temperatura entre mediciones. 4.1. Con respecto al desempeño analı́tico de la transferencia iónica La técnica de Voltamperometrı́a de Onda Cuadrada (SWV) en comparación a la Voltamperometrı́a Cı́clica (CV) permite en gran medida reducir el impacto de estas restricciones a través de mecanismos como: reducción de las corrientes de fondo a un origen capacitivo, aumento de la respuesta electroquı́mica de transferencia sobre el aumento de la corriente capacitiva (comparada a CV) y mayor facilidad de inspección y tratamiento de los voltamperogramas resultantes (lo cual reduce los errores interpretativos). La posibilidad de realizar una sustracción del fondo en SWV permite aislar la 92 señal correspondiente al proceso de transferencia, sin embargo, no es suficiente para eliminar totalmente dicho fondo, como queda evidenciado en los interceptos de las curvas de calibración obtenidas. La persistencia de estos interceptos tanto en presencia como ausencia de analito indica que es una caracterı́stica inherente a la técnica. Como resultado, existe una variabilidad importante de los resultados que se obtienen en diferentes celdas, la cual se evidencı́a por valores de R2 menores a 0.9999 en las curvas de calibracion. El ajuste lineal, sin embargo, es válido y se muestra funcional en el análisis en donde valores tı́picos para los lı́mites de confianza están en el rango de ±1µM . Los LDM y LCM obtenidos para el análisis de CH+ y TMA+ a través de la técnica de SWV mediante una transferencia simple son equiparables a aquellos reportados para la transferencia facilitada de otros iones en condiciones similares [18], [28]; más aún, son menores que los que se podrı́an obtener en la técnica de CV sin comprometer severamente el análisis debido al aumento de las corrientes de fondo. A esto se le puede sumar la facilidad y precisión de lectura del voltamperograma de SWV en contraposición al de CV lo cual mejora el desempeño analı́tico del método debido a que reduce la incertidumbre en la determinación el potencial y corriente de pico. Para el caso del TBA+ si bien los LDM y LCM son sustancialmente mayores, implican una gran ganancia en contraste con lo que se puede medir en CV. La posibilidad de desempalmar de una manera sencilla la señal de transferencia del analito de la señal de la ventana de potencial en SWV da lugar a un voltamperograma caracterizable y cuya respuesta electroquı́mica se ve notablemente mejorada por una intensidad y reproducibilidad mayor que en CV. A lo largo del estudio se llevaron a cabo notables mejoras en lo respectivo a la calidad de los instrumentos y procedimientos necesarios para llevar a cabo el experimento. El desarrollo de métodos adecuados para la obtención de electrodos Ag/AgCl y de platino, para el llenado de la celda y para la obtención de una interfase plana fueron de gran importancia para obtener mediciones reproducibles (dentro de la variación natural de la técnica). En este sentido, la calidad en el desenvolvimiento experimen- 93 tal y en las habilidades del analista son de gran importancia para asegurar lecturas reproducibles. Finalmente, el impacto de la temperatura en las mediciones fue un aspecto que se reconoció como de gran importancia ya avanzado el estudio. El reconocimiento de la transferencia de iones como un proceso activado sugiere que el control de temperatura debe ser de ± 0.1 o C. Esto no se logró en la etapa experimental primero debido a una desestimación del efecto y en una fase posterior debido a la imposibilidad de realizarlo debido a severas interferencias de los mecanismos de control y medición de temperatura con el potenciostato. Para las mediciones realizadas, este factor pudo introducir una variabilidad de ± 5 % en estas, lo cual es equiparable para concentraciones de intermedias a altas a la contribución de la señal residual de las curvas de calibración. El efecto, sin embargo, está ya bien caracterizado y mejoras o proyectos secuenciados a este deberán considerar a la temperatura como un factor de gran impacto en las mediciones. 4.2. Con respecto a la evaluación de señales y empalmes Dos procesos pueden ser bien distinguidos y caracterizados con un error mı́nimo si la separación en SWV entre sus potenciales de pico es de al menos 0.2 V. Esto queda fácilmente evidenciado cuando se evalua la transferencia simultánea de TBA+ y TMA+ . El voltamperograma resultante de la transferencia de un solo ión puede ser analizado mediante un procedimiento de linealización del potencial de celda contra una función que depende principalmente del parámetro de altura del pulso en SWV, P H. En general, para esta transferencia de un ion, se obtiene un valor de R2 tı́pico mayor a 0.99, indicando la bondad del ajuste. Cuando se presentan procesos empalmados, es decir, cuyos potenciales de transferencia difieren en menos de 0.2 V, la linealización antes descrita será deficiente y presentará valores de R2 menores a 0.99. El comportamiento de la señal 94 resultante de transferencia mixta evaluada experimentalmente es consistente con las simulaciones realizadas para el empalme de señales, mostrando que el procedimiento de linealización es una manera efectiva de determinar dichas transferencias mixtas. Si bien el método no permite discernir la identidad de las especies que se transfieren en una señal empalmada, es una base importante para asegurar la presencia de una sola especie en caso de que el ajuste sea bueno o bien, descartar esto si el ajuste es deficiente; más aún, inclusive la presencia de interferencias que se encuetren en una proporción de 1:6 con respecto a un determinado analito de interés se puede determinar mediante el método. 4.3. Con respecto al comportamiento térmico del sistema Se encontró una fuerte dependencia de la posición y la intensidad de la señal electroquı́mica con respecto a la temperatura en la evaluación por SWV. El estudio del proceso de transferencia a distintas temperaturas se prefirió por esta técnica al ser evidente que su rapidez y facilidad de lectura mejoraban notablemente el estudio del fenómeno. La variación del potencial de pico con respecto a la temperatura resulta una relación que queda bien descrita por un modelo lineal, aunque existe un grado considerable de desajuste. La relación lineal es consistente con el desplazamiento de múltiples potenciales que existen en la celda, al menos uno por cada interfase además de la interfase de estudio. Debido a este acoplamiento de potenciales, es imposible fincar el desplazamiento de una serie de señales de un ión de estudio a la entropı́a de transferencia. Una manera de evitar éste problema es obtener la diferencia entre las pendientes obtenidas a partir del ajuste lineal transformadas a unidades de entropı́a para dos iones que se transfieren en una misma celda, a manera que la contribución entrópica de los diversos potenciales acoplados se elimine. La 95 diferencia en el caso de la transferencia simultánea de TBA+ y TMA+ es consistente con lo reportado en la literatura, aunque bien inclusive en lo ya reportado hay una discrepancia considerable en los valores para distintas técnicas. La intención primera del estudio de la transferencia de dos señales empalmadas a distintas temperaturas fue la separación de estas en virtud de su diferente entropı́a de transferencia; esto no se logró debido a que la variación de los potenciales acoplados es mucho mayor a la entropı́a de transferencia de un determinado ion y por lo tanto las señales no se desempalman en un grado aceptable. Conocer la dependencia lineal para la el potencial de pico con respecto a la temperatura es importante para referenciar en una celda dada el valor obtenido con el valor a una temperatura de calibración. La variación de la intensidad de señal con respecto a la temperatura obedece en mejor medida a un modelo lineal. Esta dependencia lineal por un lado es inusitada debido a que el término de temperatura aparece en el modelo para ∆Ip bajo distintas formas: desde términos lineales hasta exponenciales. La principal componente de este desplazamiento proviene del cambio en el coeficiente de difusión Di de la especie transferida a través de la interfase. El ajuste a una función lineal es conveniente desde el punto de vista analı́tico ya que permite la corrección sencilla de datos de mediciones a distintas temperaturas para referenciarlos a las curvas de calibración obtenidas, en general, con buenos resultados. Modificar la temperatura durante el análisis de una especie puede ser benéfico debido a que a bajas temperaturas hay un achatamiendo de las corrientes de fondo, lo cual permite reconocer con mayor facilidad procesos empalmados; a temperaturas mayores, la intensidad de la señal se ve notablemente mejorada, lo cual puede ser de interés para intensificar la corriente de pico para un proceso dentro de la ventana de potencial en virtud de que la corriente capacitiva no cambia notablemente. Esta último es una aplicación interesante en el sentido de que se evitan complicaciones instrumentales como las observadas al intentar elevar el valor para P H. 96 Bibliografı́a [1] Marecek V., Samec Z. y Koryta J. “Charge Transfer Across the Interface of Two Inmiscible Electrolyte Solutions”. Advances in Colloid and Interface Science 29, 3, (1988). [2] Nernst W. y Riesenfeld E.H. Ann.Phys 8, 600, (1908). [3] Gavach C. Experientia Suppl. 18, 321,(1971). [4] Senda M., Kakiuchi T. y Osakai T. “Electrochemistry at the Interface between two inmiscible electrolyte solutions”.Electrochimica Acta 36(2), 253, (1991). [5] Reymond F., Fermin D., Lee H.J. y Girault H. “Electrochemistry at liquid/liquid interfaces: methodology and potential applications”.Electrochimica Acta 45(2), 2647, (2000). [6] Frank S. y Schmickler W. “Structure of liquid/liquid interfaces from a lattice gas model”. Journal of Electroanalytical Chemistry 564, 239, (2004). [7] Stephenson M., Holmes S. y Dryfe R. “A novel approach to the elucidation of facilitated ion transfer mechanisms at the liquid/liquid interface”.Electrochemistry Communications 6, 294, (2004). [8] Reymond F., Steyeart G., Carrupt P.A., Testa B. y Girault H. “Ionic Partition Diagrams: A Potential-pH Representation”.Journal of the American Chemical Society 118, 11951, (1996). [9] Reymond F., Chopineaux-Courtois V., Steyeart G., Bouchard G., Carrupt P.A., Testa B. y Girault H. “Ionic partition diagrams of ionisable drugs: 97 pH-lipophilicity profiles, transfer mechanisms and charge effects on solvation”.Journal of Electroanalytical Chemistry 462, 235, (1999). [10] Samec Z., Samcová E. y Girault H. “Ion amperometry at the interface between two inmiscible electrolyte solutions in view of realizing the amperometric ionselective electrode”. Talanta 63, 21, (2004). [11] Ohde H., Uehara A., Yoshida Y., Maeda K., Kihara S. “Some factors in the voltammetric measurement of ion transfer at the micro aqueous/organic solution interface”. Journal of Electroanalytical Chemistry 496, 110, (2001). [12] Reymond F. “Electrochemistry at Liquid/Liquid Interfaces to Determine Lipophilicity of Drugs and Ionisable Compounds”. Tesis, EPFL, (1998). [13] Bard A. y Faulkner L.Electrochemical Methods: Fundamentals and Applications. 2a ed. John Wiley & Sons, Inc.,Nueva York: 2001. [14] Scholz F. (Ed). Electroanalytical Methods: Guide to Experiments and Applications.Springer, Berlin: 2002. [15] Tomazsewsky L. “Assisted transfer of Heavy Metals at the Liquid/Liquid interface: Elucidation of transfer Mecanisms”.Tesis, EPFL,(2000). [16] Gobry V. “Ion Transfer of Acids and Bases”.Tesis, EPFL,(2001). [17] Mirceski V., Gulaboski R., y Scholz F. “Square-wave thin-film voltammetry: influence of uncompensated resistance and charge transfer kinetics”.Journal of Electroanalytical Chemistry 566, 351, (2004). [18] Sherburn A., Arrigan D., Dryfe R. y Boag N. “Square-Wave Voltammetric Transfer of Silver Ions Across the Water/1,2-dicloroethane Interface”.Electroanalysis 16(15), 1227, (2004). [19] Covello M. y Schettino O. “Applicazione della cromatografı́a su carta e su strato sottile alla recerca degli antifermentativi negli alimenti, Nota IV- Identificazione e dosaggio dei Sali ammonici quaternari”. Il Farmaco.Ed.Pr. 21(3), 145, (1967). 98 [20] Arugonda S.K. “Quaternary Ammonium Compounds”. International Programme on Chemical Safety, Poisons Information Monograph G022. Julio de 1999. http://www.inchem.org/documents/pims/chemical/pimg022.htm. [21] Rounbehler D., Achter E., Fine D., Freeman F., MacDonald S. y Klotzsch H. “Method and system for sampling and determining the presence of salts of ammonia and amines in containers”. Patente US5,418,170. 23 de mayo de 1995. [22] Rasulev U.K., Nazarov E.G. y Khudaeva G.B. “Chromatoghraphic determination of trace amounts of amines using a surface ionization detector”.Journal of Chromatography A 704, 473, (1995). [23] Bourchard G., Carrupt P.A., Testa B., Gobry V. y Girault H. “The apparent lipophilicity of Quaternary Ammonium ions is influenced by Galvani Potencial Difference, not Ion-Pairing: A cyclic voltammetry study”.Pharmaceutical Research 18(5), 702, (2001). [24] Rahman A. y Doe H. “Ion transfer of tetraalkylammonium cations at an interface between frozen aqueous solution and 1,2-dichloroethane”.Journal of Electroanalytical Chemistry 424, 159, (1997). [25] Martins M.C., Pereira C.M., Girault H.H y Silva F. “Specific adsorption of tetraalkylammonium cations on the 1,2-dichloroethane/water interface”.Electrochimica Acta 50, 135, (2004). [26] Ding Z. “Spectroelectrochemistry and photoelectrochemistry of charge transfer at liquid/liquid interfaces”. Tesis, EPFL,(1999). [27] Princeton Applied Research. Technical Note 101 [28] Beni V., Ghita M. y Arrigan D. “Cyclic and pulse voltammetric study of dopamine at the interface between two inmiscible electrolyte solutions”. Biosensors & Biolectronics20, 2097, (2005). 99 [29] Samec Z., Lhotsky A., Jänchenová H., y Marecek, V. “Interfacial tension and impedance measurements of interfaces between two inmiscible electrolyte solutions”. Journal of Electroanalytical Chemistry43, 47, (2000). [30] Day R.A. y Underwood A.L. Quı́mica Analı́tica Cuantitativa,5o ed. Prentice-Hall, México, 1998. 45-48. [31] Aoki K., Maeda K. y Osteryoung J. “Characterization of Nernstian square-wave voltammograms”. Journal of Electroanalytical Chemistry 272, 17, (1989). [32] Hefter G., Marcus Y. y Waghorne W.E. “Enthalpies and Entropies of Transfer of Electrolytes and Ions from Water to Mixed Aqueous Organic Solvents”. Chemical Reviews 102, 2773, (2002). [33] Osakai T., Ogawa H., Ozeki T. y Girault H. “Determination of the Entropy of Ion Transfer between Two Inmiscible Liquids using the Water/Oil/Water Thermocouple”.Journal of Physical Chemistry B107, 9829, (2003). 100