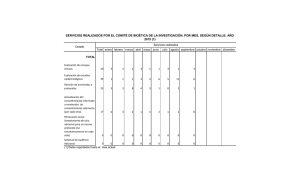
Autonomía e investigación clínica 2010
Anuncio

colección MATERIALES DE ÉTICA APLICADA A LA INTERVENCIÓN SOCIAL, 2 C M Aspectos éticos y jurídicos de la investigación clínica con personas adultas incapaces. Guía de análisis para los Comités Éticos de Investigación Clínica Y CM JOAN CANIMAS i BRUGUÉ (coord.) CY CMY K con el patrocinio de: con la colaboración de: Autonomía e Investigación Clínica 2010 MY colección MATERIALES DE ÉTICA APLICADA A LA INTERVENCIÓN SOCIAL, 2 observatori d’ètica aplicada a la intervenció social C A M P U S A R N A U D ’ E S C A L A Innovació i Recerca Social i Sociosanitària Autonomía e investigación clínica 2010 Aspectos éticos y jurídicos de la investigación clínica con personas adultas incapaces. Guía de análisis para los comités éticos de investigación clínica Autonomía e investigación clínica 2010 Aspectos éticos y jurídicos de la investigación clínica con personas adultas incapaces. Guía de análisis para los comités éticos de investigación clínica Joan Canimas Brugué (coord.) Colección MATERIALES DE ÉTICA APLICADA A LA INTERVENCIÓN SOCIAL, 2 OBSERVATORI D’ÈTICA APLICADA A LA INTERVENCIÓ SOCIAL FUNDACIÓ CAMPUS ARNAU D’ESCALA Parc Científic i Tecnològic de la UdG Edificio Jaume Casademont, puerta B, despacho 9 Pic de Peguera, 15, (la Creueta) 17003 Girona Teléfono: 972 10 42 15 http://etica.campusarnau.org Coordinación editorial: Joan Canimas, Sílvia Monserrat i Eduard Solé Revisión de los textos: Servei de Llengües Modernes de la UdG © Diseño del interior, maquetación y corrección: Editorial Glosa, S.L. Avinguda de Francesc Cambó, 21, 5ª planta – 08003 Barcelona Teléfonos: 932 684 946 / 932 683 605 – Telefax: 932 684 923 www.editorialglosa.es ISBN: 978-84-7429-511-5 Dipósito legal: © Diseño de portada: Ramon Vilageliu © Texto: Joan Canimas i Brugué © Distribución y explotación: Fundació Campus Arnau d’Escala Reservados todos los derechos. Ninguna parte de esta publicación puede ser reproducida ni transmitida en ningún formato o medio, incluidas las fotocopias o cualquier otro sistema de recuperación de almacenamiento de información, sin la autorización por escrito del titular de los derechos. Esta edición ha sido posible gracias a PFIZER y a IAS. 5 ÍNDICE INTRODUCCIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15 PRÓLOGO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15 METODOLOGÍA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 ADVERTENCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 RECOMENDACIONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 I. CONDICIONES CIENTÍFICAS, ADMINISTRATIVAS Y SOCIALES . . . . . . . 19 1 La investigación clínica debe perseguir una mejora médica, estar científicamente justificada y bien planteada, y regirse por el criterio de conseguir el máximo beneficio con los mínimos riesgos y molestias posibles para el paciente. El equipo investigador debe tener las aptitudes y condiciones necesarias para realizar la investigación . . . . . . . . . . . . . . . . . . 19 2 La investigación clínica con personas que no tienen plena capacidad de dar el consentimiento no debe poder hacerse, con eficacia comparable, con pacientes capaces de darlo, y debe estar relacionada directamente con algún proceso clínico que sufran las personas a las cuales va dirigida . . . . . . 20 3 En la reunión del CEIC debe participar un experto en la patología tratada y en el grupo de pacientes afectados por el estudio clínico, o se le debe haber pedido asesoramiento . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20 4 En la reunión del CEIC tendría que participar un representante de la asociación o asociaciones de personas afectadas por la patología estudiada . . . . . . . . . . . . . . . . . . . . . 20 5 El CEIC debe prestar atención a que no se produzca una «sobreutilización» de algún perfil social o económico en el reclutamiento de pacientes si no es por un criterio científico éticamente justificable . . . . . . . . . . . . . . . . . . . . . . . . 22 6 II. VALORACIÓN DE LOS BENEFICIOS, RIESGOS Y MOLESTIAS . . . . . . . 23 6 La minimización o ausencia de riesgo no es una condición suficiente para garantizar el respeto a las personas que participarán en un estudio clínico. También hay que ponderar las posibles molestias, es decir, el dolor, las incomodidades, el miedo, la coerción o cualquier perturbación del bienestar o la tranquilidad de la persona . . . . . . . . . . . . . . . . . . . . . . . . . . 23 7 La investigación no debe implicar riesgos ni molestias desproporcionados en relación con los beneficios potenciales que se puedan obtener. El protocolo del estudio clínico tendría que valorar y justificar la relación entre los beneficios y los riesgos y molestias; el CEIC debería validar o corregir esta valoración (después de su propio análisis), y la hoja de información debería informar al paciente, tutor, curador o guardador de hecho . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 8 En la valoración de estudios clínicos hay que estar alerta a la maximización de los beneficios y la minimización de los riesgos que puede provocar el diferente ámbito de afectación (ciencia y futuros pacientes por un lado, paciente que participa en la investigación por el otro) . . . . . . . . . . . . . . 27 9 Los beneficios, riesgos y molestias del estudio clínico deben compararse con los beneficios, riesgos y molestias del tratamiento convencional. Y si no hay tratamiento convencional, con los beneficios, riesgos y molestias que tendría el paciente si no recibiera ningún tratamiento y una correcta atención sanitaria . . . . . . . . . . . . . . . . . . . . . . . . 29 10 El protocolo del estudio clínico tendría que diferenciar los beneficios y los riesgos y molestias directamente asociados al tratamiento experimental con posibles beneficios directos (R-MBD) de los beneficios y riesgos y molestias derivados de observaciones complementarias con beneficios no directos que persiguen aumentar el conocimiento científico (R-MBND), si los hay . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 7 11 El CEIC debería evaluar y aprobar, por un lado, el tratamiento experimental y, por el otro y si las hay, las pruebas complementarias que persiguen aumentar el conocimiento científico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32 12 Los estudios clínicos dirigidos a personas incapaces de otorgar el consentimiento informado en los que los beneficios directos esperados son inferiores a los riesgos y molestias previstos (BD < R-M) o de los que no se prevén beneficios directos y sí riesgos y molestias (R-MBND), sólo deberían autorizarse cuando: (a) uno de los criterios de reclutamiento sea que el paciente haya autorizado, antes de la incapacidad y a través de un documento de instrucciones previas, participar en este tipo de estudios, o (b) el tipo de patología no haga posible promocionar las instrucciones previas en este tipo de investigación (p. ej., discapacidad intelectual congénita o traumatismo craneoencefálico). En esta última situación, la autorización debe ser muy prudente y exigente . . . . . . . . . 33 13. La inclusión en estudios clínicos de personas que se encuentran en una situación clínica de emergencia sin capacidad (temporal o permanente) de dar el consentimiento sólo se debería autorizar cuando, además de los requisitos señalados en la recomendación 12, haya probabilidades fundamentadas de que si participa en ellos obtendrá beneficios terapéuticos que no se pueden obtener de ningún otro modo. En el supuesto de estudios clínicos sin beneficios directos (BND), las condiciones deberían ser las mismas que las señaladas en la recomendación 12 de esta guía . . . . . . . . . 45 III. ESTUDIOS CON PLACEBO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 14 En investigaciones clínicas con personas adultas incapaces o que no tienen plena capacidad de otorgar el consentimiento informado, sólo es aceptable que el grupo control tome placebo o no reciba ningún tratamiento en estudios para los cuales no hay tratamiento convencional de eficacia probada o, en caso de que lo haya, no tiene eficacia en los posibles participantes en el estudio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 8 IV. CAPACIDAD DEL PACIENTE PARA TOMAR DECISIONES . . . . . . . . . . 51 15 Evaluar la competencia de una persona para dar el consentimiento es una tarea de mucha responsabilidad profesional y moral. Hay que evaluar cada caso concreto utilizando procedimientos reconocidos, la pericia profesional y, cuando haga falta, la decisión participada . . . . . . . . . . . . . . . 51 16 La necesidad de valorar con precisión la capacidad del paciente para dar el consentimiento aumenta a medida que lo hacen los riesgos y las molestias de la investigación clínica, especialmente cuando el estudio no aporta ningún beneficio directo para el paciente. Cuanto más elevados son los riesgos y las molestias, más elevado debe ser el grado de protección del paciente y de control de la investigación . . . . . . . . . . . . . . . . . . . . . . . . . 59 17 Ética y jurídicamente, la capacidad de decisión de una persona siempre se presupone, la incapacidad debe demostrarse y en caso de duda prevalece la presunción de capacidad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61 V. INFORMACIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61 18 El protocolo del estudio clínico debe recoger y garantizar que se dará una información oral sin prisas, veraz, precisa, exhaustiva, comprensible, continuada y adaptada a las personas a las cuales va dirigida y con un intervalo de tiempo razonable para tomar la decisión. Hay que tener especial cuidado con la capacidad de comprensión de los pacientes y también con la del tutor, curador o guardador de hecho, sobre todo si son de otros contextos culturales, y asegurarse de que han entendido las características del estudio y las consecuencias de participar en él. Si la persona que tiene que dar el consentimiento tiene dificultades para entender la lengua con la cual el investigador le informa, es necesario que haya un traductor imparcial. Con personas de contextos culturales muy diferentes del occidental, haría falta la presencia de un mediador cultural . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61 9 19 Es necesario informar oralmente y por escrito, y siempre que sea procedente, de las treinta y siete cuestiones que se señalan en esta recomendación, algunas de las cuales se amplían en recomendaciones aparte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64 20 La hoja de información para el paciente debe estar redactada de manera veraz, precisa, exhaustiva, comprensible y adaptada a las capacidades de las personas a las que va dirigida . . . . . . 68 21 Además de la hoja de información dirigida a los tutores, a los pacientes con dificultades de comprensión, apreciación y razonamiento debe facilitárseles una hoja de información resumida de fácil comprensión o cualquier otro apoyo informativo adaptado a sus capacidades, a fin de que puedan intervenir tanto como sea posible en la toma de decisiones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69 22 En estudios clínicos con riesgos y molestias derivados de un tratamiento experimental con posibles beneficios directos (R-MBD), pero también con riesgos y molestias de pruebas complementarias sin beneficios directos para el paciente (R-MBND), debería disponerse de una hoja de información y de un consentimiento por escrito para el tratamiento experimental y otro para las pruebas complementarias . . . . . 70 23 Si se pretende hacer un análisis genético, es necesario proporcionar información expresa y específica y el consentimiento correspondiente . . . . . . . . . . . . . . . . . . . . . . . . 70 24 Quien firma el consentimiento informado no debe tener ningún tipo de duda sobre el hecho de que puede negarse a participar en la investigación y retirarse en cualquier momento sin necesidad de dar explicaciones y sin que eso repercuta de ninguna manera en su asistencia y relación sanitaria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71 10 25 La información oral y escrita debería advertir que hay que prestar atención a las posibles relaciones de dependencia del paciente, tutor, curador o guardador de hecho con el profesional sanitario que les propone participar en la investigación, a fin de que esta situación no influya en la decisión . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71 26 Cuando no se espera ningún beneficio directo para el paciente, en el proceso de información oral hay que garantizar que las personas afectadas no tienen una «falsa idea terapéutica» o «equívoco terapéutico» (therapeutic misconception) . . . . . . 72 27 Cuando el beneficio directo previsto sea igual o menor que los riesgos y molestias y el consentimiento es por representación, la información oral y escrita debería advertir que hay que prestar atención en la influencia que puede tener la posible desazón de impotencia ante la enfermedad o incluso de deuda con el paciente, y recordar la necesidad de una decisión adecuada a las circunstancias y proporcionada a las necesidades del paciente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73 28 Los pacientes, tutores, curadores o guardadores de hecho deberían disponer, además, de una guía que informe de lo que significa participar en un estudio clínico y que les oriente y facilite el proceso de decisión . . . . . . . . . . . . . . . . . . . . . . . . . 73 29 Las asociaciones de afectados deberían estar informadas de los estudios clínicos que se realizan y ofrecer un servicio de apoyo a las personas que se plantean participar en ellos . . 75 30 No se puede dar ningún incentivo o estímulo económico; sólo se pueden retribuir los gastos generados por participar en la investigación. Si en el transcurso del estudio se da algún regalo, en ningún caso puede tener un valor persuasivo y este gesto debería ser aprobado por el CEIC . . . . . . . . . . . . . . . . . . . . . . 75 11 VI. CONSENTIMIENTO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76 31 Cuando el paciente está incapacitado legalmente, debe obtenerse autorización de su representante legal. El paciente debe intervenir tanto como sea posible en el proceso informativo y en la toma de decisiones. Debe respetarse su rechazo o asentimiento y si su capacidad no permite expresarlo, debe tenerse en cuenta su presunta voluntad . . . . . . . . . . . . . . 76 32 Cuando el paciente está incapacitado legalmente no siempre prevalece la decisión del tutor sobre la participación o no en el estudio clínico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 33 Cuando el paciente está incapacitado legalmente, tiene una patología que no ha hecho posible la manifestación de instrucciones previas y no se prevén beneficios directos del estudio clínico (BND) y sí riesgos y molestias, antes de incluirlo habría que indagar si la autorización se da en un contexto de cuidado y estima al paciente y si manifestó voluntades anticipadas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80 34 Cuando los riesgos y las molestias de un tratamiento médico experimental en personas incapacitadas legalmente son muy elevados, es necesaria la autorización judicial . . . . . . . . . . . . . 82 35 Cuando el paciente no es plenamente competente y no ha sido incapacitado legalmente, debe obtenerse también la autorización de las personas que le cuidan, aunque eso hoy no esté previsto legalmente . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82 36 Cuando el paciente no es plenamente competente y no ha sido incapacitado legalmente, debería advertirse que, como criterio general, prevalece la decisión del paciente y que en los casos en que se considera que no es prudente hacerlo, debe intervenir el juez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84 37 Si la persona que ha de dar el consentimiento no sabe leer, es deseable que haya un testigo imparcial (respecto al estudio clínico) en el momento de firmar el consentimiento . . . . . . . . 88 12 38 En la hoja de consentimiento por escrito deberían figurar, además de los 12 ítems para el paciente y de los 14 para el tutor actualmente establecidos, los riesgos a los que se somete el paciente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88 39 Hay que promover las instrucciones previas también en temas de investigación . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90 40 No siempre es recomendable atender las instrucciones previas en lo referente a la participación en estudios clínicos . . . . . . . 92 VII. SEGUIMIENTO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92 41 El CEIC debe establecer procedimientos para supervisar que se cumplen las condiciones y procedimientos establecidos en el protocolo del estudio clínico . . . . . . . . . . . . . . . . . . . . . . . 92 42 Cuando se producen cambios en las condiciones o procedimientos de un estudio clínico y también periódicamente en estudios a largo plazo, el investigador debe pedir de nuevo el consentimiento informado de los pacientes o tutores . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93 BIBLIOGRAFÍA, DECLARACIONES Y LEGISLACIÓN . . . . . . . 95 PERSONAS QUE HAN ELABORADO O COLABORADO EN ESTA GUÍA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101 «Aquí me tienes» 15 INTRODUCCIÓN PRÓLOGO Hasta hoy, los Comités Éticos de Investigación Clínica (CEIC) no disponen de una guía que les facilite la valoración y la autorización o rechazo de los protocolos de investigación clínica dirigidos a personas adultas incapaces o que no tienen plena capacidad para otorgar el consentimiento informado. Esta guía consta de cuarenta y dos recomendaciones, argumentadas con referentes éticos, jurídicos, conceptuales y procedimentales, que pretenden ofrecer pautas de deliberación y elementos de reflexión. Las recomendaciones están agrupadas en siete capítulos que siguen un cierto orden procedimental. Hay que advertir, sin embargo, que no todas las recomendaciones se circunscriben al proceso de valoración de los CEIC, aunque tienen que ver con ello. Se tratan también algunos aspectos que el CEIC debe tener en cuenta, pero que ya no le corresponden; por ejemplo, valorar si una persona tiene capacidad para otorgar el consentimiento para participar en un estudio clínico o no la tiene (recomendaciones 15 y 16) o, en caso de que no tenga esta capacidad, decidir si prevalece o no su opinión (recomendaciones 32 y 33). Las recomendaciones más controvertidas (por ejemplo, la 12) son más prolíficas en argumentaciones que aquellas otras en las que hay un consenso generalizado que ha quedado plasmado en la ley o en los protocolos de buenas prácticas clínicas. Algunas recomendaciones de esta guía proponen pautas de actuación o análisis que no están recogidos en la normativa vigente o en la mayoría de protocolos. De éstas, consideramos que hay que destacar las siguientes: 1) que en la reunión del CEIC haya un representante de la asociación o asociaciones de afectados por la patología estudiada (recomendación 4); 2) que el protocolo del estudio clínico no describa simplemente los beneficios y los riesgos y molestias previstos, sino que valore la relación que hay entre ellos (recomendación 7); 3) la necesidad de diferenciar los riesgos y molestias directamente asociados al tratamiento experimental con posibles beneficios directos, de 16 los riesgos y molestias derivados de observaciones complementarias sin beneficios directos que persiguen aumentar el conocimiento científico, si los hay (recomendaciones 10 y 22); 4) que los protocolos actuales y la legislación vigente no protegen suficientemente la dignidad y los derechos de las personas incapaces o que no tienen plena capacidad de otorgar el consentimiento informado que participan en ensayos clínicos de los cuales no se prevén beneficios directos y sí riesgos y molestias, o los beneficios directos esperados son inferiores a los riesgos y molestias previstos, o son ensayos clínicos pensados para situaciones de urgencia clínica (recomendaciones 12 y 13), ante lo cual se proponen algunas medidas procedimentales y legislativas para corregir esta situación; 5) se hace una primera aproximación a una taxonomía de la relación entre beneficios y riesgos y molestias (recomendación 7), que después se utiliza en unas tablas orientativas para la toma de decisiones (recomendaciones 32 y 36). De este proceso analítico hay que destacar también el hecho de tener en cuenta los diferentes «ámbitos de afectación» de la investigación a la hora de medir y comparar los beneficios y riesgos y molestias previstos (recomendación 8). Consideramos que los instrumentos conceptuales que proponemos facilitan el análisis y la valoración ética. METODOLOGÍA Para elaborar esta guía se ha utilizado la metodología siguiente: 1) Cuatro sesiones de trabajo de un grupo de expertos (Joan Canimas Brugué, Gemma Camps Rovira, Joan M. del Pozo Àlvarez, Jordi Ferrer Beltran y Joan Vilalta Franch). 2) Redacción del documento marco (Joan Canimas Brugué) y revisión por parte del grupo de expertos. 3) Lecturas de contraste (Josep Garre Olmo, Secundí López-Pousa, Sílvia Monserrat Vila, José Luis Molinuevo Guix y Francesc Xavier Pereda Gámez). 4) Redacción del documento final (Joan Canimas Brugué). 17 ADVERTENCIAS 1) Las recomendaciones con la forma verbal de indicativo («es», «debe», «no se puede»...) son recomendaciones recogidas en la normativa vigente o ampliamente aceptadas, y las que utilizan la forma condicional («habría que…») son propuestas de nuevas pautas de actuación, algunas de las cuales implican cambios en la legislación vigente o en los protocolos de buenas prácticas. 2) La literatura científica, influida por la distinción norteamericana entre capacity (un término de significación psicológica y clínica) y competency (un término de significación jurídica), suele diferenciar entre capacidad y competencia. Para facilitar el análisis y para evitar esta diferenciación, siempre confusa,1 en esta guía se habla de personas adultas incapaces de otorgar el consentimiento informado para participar en un estudio clínico, que pueden estar incapacitadas legalmente —entonces hablaremos de «personas incapacitadas legalmente»— o no —entonces hablaremos de «personas incapaces»—. Pero como, tal y como se señala diversas veces a lo largo de estas páginas, la capacidad de decidir de una persona no es siempre un rasgo dicotómico («personas capaces» o «personas incapaces»), ni los conceptos «personas incapacitadas legalmente» y «personas incapaces» pueden abarcar toda la variedad de situaciones, nos ha parecido necesario referirnos a las personas adultas sin plena capacidad de otorgar el consentimiento informado para participar en un estudio clínico —entonces hablaremos de «personas sin plena capacidad»—. Aunque este término a veces introduce un factor de incertidumbre, consideramos que no está reñido con el aumento de precisión, en tanto que capta un abanico de personas difícilmente incluidas en las categorías «personas capaces», «personas incapacitadas legalmente» y «personas incapaces». La recomendación 15 intenta delimitar estos contornos. 1 Sobre la cuestión terminológica de los conceptos capacity, competency, capacidad y competencia, véase SIMÓN-LORDA, P. (2008): «La capacidad de los pacientes para tomar decisiones: una tarea todavía pendiente», Revista de la Asociación Española de Neuropsiquiatría, 2008, vol. XXVIII, n.º 102, p. 325-348. 18 3) Con respecto a las figuras que ejercen la guarda y la protección de las personas adultas incapaces o sin plena capacidad de otorgar el consentimiento informado, los artículos 215, 216 y 227 del Código civil hablan de tutor, curador, defensor judicial, guarda de hecho o de derecho y de administrador. Las tres figuras a las cuales recurriremos en esta guía son el tutor, el curador y el guardador de hecho. 19 RECOMENDACIONES I. CONDICIONES 1 CIENTÍFICAS, ADMINISTRATIVAS Y SOCIALES La investigación clínica debe perseguir una mejora médica, estar científicamente justificada y bien planteada, y regirse por el criterio de conseguir el máximo beneficio con los mínimos riesgos y molestias posibles para el paciente. El equipo investigador debe tener las aptitudes y condiciones necesarias para realizar la investigación 1.1 Rigor científico. Según la legislación vigente, los estudios clínicos deben basarse en el conocimiento científico disponible, y la información buscada debe suponer, previsiblemente, un avance en el conocimiento científico sobre el ser humano, o deben mejorar su estado de salud (pertinencia del estudio). Sólo se pueden hacer cuando se dispone de suficientes datos científicos y, en particular, ensayos farmacológicos y toxicológicos que garanticen que los riesgos que implica para la persona a la cual se realiza son admisibles. La investigación debe estar bien diseñada y tener en cuenta los últimos avances científicos, y el equipo investigador las aptitudes y condiciones necesarias.2 1.2 Minimizar los riesgos. La legislación también señala que hay que minimizar los riesgos para las personas que participan en estudios, reducir al mínimo el dolor, la incomodidad, el miedo y cualquier otro riesgo previsible, especialmente cuando los sujetos del ensayo sean menores, adultos incapaces o constituyan una población especialmente vulnerable a causa de su situación económica, médica o social.3 2 Pauta 1 de las Pautas éticas internacionales para la investigación biomédica en seres humanos preparadas por el Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS) en colaboración con la Organización Mundial de la Salud (OMS) (2002).Art. 3 del RD 223/2004 de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. Art. 8 del Protocolo adicional al Convenio sobre los Derechos humanos y la biomedicina, relativo a la investigación biomédica (2005). Art. 4 de la Declaración universal sobre bioética y derechos humanos (2005). Arts. 10 y 16 de la Ley española 14/2007, de 3 de julio, de investigación biomédica.Art. 12 de la Declaración de Helsinki (2008). 3 Art. 3.5 del Real decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. 20 2 La investigación clínica con personas que no tienen plena capacidad de dar el consentimiento no debe poder hacerse, con eficacia comparable, con pacientes capaces de darlo, y debe estar relacionada directamente con algún proceso clínico que sufran las personas a las cuales va dirigida 2.1 Según la legislación vigente, la investigación con personas sin plena capacidad para dar el consentimiento sólo puede hacerse cuando el estudio es esencial para validar datos obtenidos en ensayos clínicos efectuados con personas sanas capaces de otorgar el consentimiento informado (fase I de un estudio clínico), cuando el estudio tiene relación directa con la enfermedad que sufre la persona y cuando no se puede hacer con eficacia equiparable con sujetos capaces de dar el consentimiento.4 3 En la reunión del CEIC debe participar un experto en la patología tratada y en el grupo de pacientes afectados por el estudio clínico, o se le debe haber pedido asesoramiento 3.1 Según la legislación vigente, en el proceso de aprobación de un protocolo de investigación que incluya mayores de edad incapacitados legalmente, es imprescindible la presencia de un experto en la enfermedad en cuestión o haber pedido asesoramiento sobre las cuestiones clínicas, éticas y psicosociales en el ámbito de la enfermedad y del grupo de pacientes afectados.5 4 En la reunión del CEIC tendría que participar un representante de la asociación o asociaciones de personas afectadas por la patología estudiada 4.1 La legislación actual no prevé esta posibilidad, aunque sería recomendable que lo hiciera. La legislación actual no prevé la presencia en el CEIC de ningún representante de asociación o asociaciones de personas afectadas por la patología sobre la cual se valora 4 Norma 4.8.14 de la Guía de buena práctica clínica CPMP/ICH/95. Art. 5 del RD 223/2004. Art. 15.ii del Protocolo adicional al Convenio para la protección de los derechos humanos (2005). Pauta 15 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). Art. 7 de la Declaración universal sobre bioética y derechos humanos (2005). Art. 20.1.b de la Ley española 14/2007. 5 Art. 5.g de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo relativa a la aplicación de buenas prácticas clínicas en la realización de ensayos clínicos con medicamentos. 21 la conveniencia o no de aprobar el protocolo de investigación.6 Consideramos recomendable que en la deliberación de los CEIC se incorporen uno o dos representantes de las asociaciones de enfermos de la patología abordada por el protocolo de investigación,7 lo cual facilitaría una visión más amplia de la cuestión, llegar a consensos con las personas que viven el problema de cerca y dar un paso más en la recomendación del artículo 18 de la Declaración universal de bioética y derechos humanos, que dice que hay que promover la transparencia en la adopción de decisiones, establecer un diálogo permanente entre las personas y los profesionales interesados y la sociedad en conjunto, y promover un debate público pluralista e informado, en el cual se expresen todas las opiniones pertinentes. 4.2 Pluralidad y representación ciudadana en los comités. El comentario sobre la pauta 2 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002) dice lo siguiente sobre la composición de los comités de ética: «Los comités de evaluación ética nacionales o locales deberían estar compuestos de manera tal que fueran capaces de proporcionar una evaluación completa y adecuada de las propuestas de investigación presentadas. Por lo general, se considera que deberían incluir médicos, científicos y otros profesionales como enfermeras, abogados, eticistas y religiosos, así como legos calificados para representar los valores culturales y morales de la comunidad y asegurar que los derechos de los sujetos serán respetados. Deberían incluir hombres y mujeres. Cuando personas sin educación o analfabetas sean foco de un estudio, deberían, asimismo, ser consideradas para formar parte del comité o invitadas para expresar sus puntos de vista. [...] Un comité de evaluación ética nacional o local, responsable de la evaluación y aprobación de propuestas para investiga- 6 Art. 12 del Real decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. En Cataluña, el art. 2.1 del Decreto 406/2006, de 24 de octubre, por el cual se regulan los requisitos y el procedimiento de acreditación de los Comités Éticos de Investigación Clínica. 7 Esto ya se está haciendo en los EE.UU.Véase ARRIOLA MANCHOLA, E.; IBARZÁBAL ARAMBERRI, X. (2007): «Fundamentos éticos de respeto al enfermo», en MARTÍNEZ LAGE, J.M.; CARNERO PARDO, C. (editores): Alzheimer 2007: recapitulación y perspectivas, Madrid, Laboratorios Andrómaco, 2007, p. 283-284. 22 ciones patrocinadas externamente, debería tener entre sus miembros o consultores a personas que estén familiarizadas con las costumbres y tradiciones de la población o comunidad en cuestión, y que sean sensibles a asuntos relativos a la dignidad humana.» Existe el peligro de que una lectura restringida del artículo 12 del Real Decreto 223/2004, que regula la composición de los CEIC, dé como resultado unos CEIC con poca pluralidad ciudadana y mucha endogamia profesional. La participación de un representante de las asociaciones de enfermos en la deliberación del CEIC, respecto a investigaciones que les afectan, facilitaría que estas asociaciones estuvieran informadas de los estudios clínicos que se realizan en su ámbito de influencia, lo cual permitiría también que las asociaciones pudieran dar apoyo a las personas que se encuentran en la situación de tener que autorizar o negar la participación en un estudio clínico (véase la recomendación 29). 5 El CEIC debe prestar atención a que no se produzca una «sobreutilización» de algún perfil social o económico en el reclutamiento de pacientes si no es por un criterio científico éticamente justificable 5.1 Del mismo modo que no hay que privar a ninguna persona ni enfermedad de los beneficios de la investigación, la equidad también requiere que ningún grupo social o económico soporte una carga de participación en la investigación clínica superior a la que le corresponde en una distribución justa. Por lo tanto, no es suficiente que el CEIC preste atención a los criterios científicos de reclutamiento, sino que también debe tener en cuenta los factores que pueden provocar un reclutamiento desproporcionado («sobreutilización» o «sobreexplotación») de algún perfil social o económico de pacientes, por ejemplo de personas de clases socioeconómicas bajas o que viven en residencias, sin que sea un criterio científica y éticamente justificado.8 8 Comentario sobre la pauta 12 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). 23 II. VALORACIÓN 6 DE LOS BENEFICIOS, RIESGOS Y MOLESTIAS La minimización o ausencia de riesgo no es una condición suficiente para garantizar el respeto a las personas que participarán en un estudio clínico. También hay que ponderar las posibles molestias, es decir, el dolor, las incomodidades, el miedo, la coerción o cualquier perturbación del bienestar o la tranquilidad de la persona 6.1 En un estudio clínico no es suficiente valorar los riesgos. También deben considerarse las posibles molestias, que incluyen el dolor, las incomodidades, la coerción o cualquier perturbación del bienestar o la tranquilidad de la persona.9 La minimización o ausencia de riesgo, por lo tanto, no es una condición suficiente para garantizar el respeto a las personas que participarán en una investigación. 7 La investigación no debe implicar riesgos ni molestias desproporcionados en relación con los beneficios potenciales que se puedan obtener. El protocolo del estudio clínico tendría que valorar y justificar la relación entre los beneficios y los riesgos y molestias; el CEIC debería validar o corregir esta valoración (después de su propio análisis), y la hoja de información debería informar al paciente, tutor, curador o guardador de hecho 7.1 Una primera clasificación de la relación entre beneficios y riesgos-molestias. La valoración de la relación entre los beneficios (directos o no directos) y los riesgos y molestias de un estudio clínico es compleja, y lo es por cuatro razones principales: i) por el grado de incertidumbre de toda investigación clínica; ii) porque deben compararse unidades diferentes (beneficio, riesgo, molestias); 9 El artículo 3.5 del Real decreto 223/2004 habla de incomodidad y de miedo («El ensayo clínico debe estar diseñado para reducir al mínimo posible el dolor, la incomodidad, el miedo y cualquier otro riesgo previsible en relación con la enfermedad y edad o grado de desarrollo del sujeto»). El artículo 7.b de la Declaración universal sobre bioética y derechos humanos (2005) habla de coerción («Las actividades de investigación que no entrañen un posible beneficio directo para la salud se deberían llevar a cabo únicamente de modo excepcional, con las mayores restricciones, exponiendo a la persona [que no tiene capacidad de dar su consentimiento] únicamente a un riesgo y una coerción mínimos»). El comentario sobre la pauta 2 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002) dice: «Las evaluaciones científica y ética no pueden estar separadas: la investigación en seres humanos científicamente inadecuada es de hecho no ética, ya que puede exponer a los sujetos a riesgos o inconvenientes sin ningún propósito; aun cuando no haya riesgo de daño, la pérdida de tiempo de los sujetos y de los investigadores en actividades improductivas representa la pérdida de un recurso valioso.» 24 iii) porque en la determinación del valor de estas unidades (beneficio, riesgo, molestias) intervienen factores subjetivos; y iv) porque el ámbito de afectación de los beneficios y de los riesgos y molestias puede ser el mismo o no (véase la recomendación 8). La relación entre beneficios y riesgos y molestias puede tener muchos valores. Una primera taxonomía, la más simple de todas, puede representarse de esta manera: TABLA 1 BD > R-M Los beneficios directos esperados (BD) son superiores a los riesgos y molestias previstos (R-M). BD ≈ R-M Los beneficios directos esperados (BD) son equiparables a los riesgos y molestias previstos (R-M). BD < R-M Los beneficios directos esperados (BD) son inferiores a los riesgos y molestias previstos (R-M). BND > R-M Los beneficios no directos10 esperados (BND), es decir, los que repercuten únicamente en el progreso de la ciencia y en futuros tratamientos, son superiores a los riesgos y molestias previstos (R-M). BND ≈ R-M Los beneficios no directos esperados (BND) son equiparables a los riesgos y molestias previstos (R-M). BND < R-M Los beneficios no directos esperados (BND) son inferiores a los riesgos y molestias previstos (R-M). BDyND > R-M Los beneficios directos y no directos esperados (BDyND) son superiores a los riesgos y molestias previstos (R-M). BDyND ≈ R-M Los beneficios directos y no directos esperados (BDyND) son equiparables a los riesgos y molestias previstos (R-M). BDyND < R-M Los beneficios directos y no directos esperados (BDyND) son inferiores a los riesgos y molestias previstos (R-M). 10 A veces también se utiliza «beneficios indirectos». Aunque no hay duda de que los dos términos son sinónimos, hemos preferido «beneficios no directos» porque nos parece que refuerza la idea de que hay beneficios a pesar de que el paciente no reciba ninguno. 25 En todo estudio clínico con posibles beneficios directos (BD) también se obtendrán beneficios no directos (BND), porque se validarán o refutarán hipótesis que beneficiarán a futuros pacientes. Sin embargo, en estos tipos de estudio es necesario valorar por separado el beneficio que repercute únicamente en el paciente que participa en la investigación (BD). La distinción entre investigación terapéutica (que supone un beneficio directo para el paciente que se somete a la experimentación) e investigación no terapéutica (sin beneficio individual directo) que establecía la Declaración de Helsinki en la versión de 1964, ha dejado de utilizarse y ha sido sustituida por la consideración y ponderación de la relación entre los beneficios y los riesgos y molestias. Se ha alegado que, en un sentido estricto, ninguna investigación puede ser considerada terapéutica, precisamente porque se encuentra en una fase de experimentación para determinar si lo es o no.Ahora bien, continúa siendo imprescindible diferenciar entre estudios clínicos con beneficios personales o directos para el paciente (BD) y estudios clínicos con beneficios no directos (BND). 7.2 Sopesar la relación beneficios/riesgos-molestias. En investigación clínica hay dos cuestiones básicas: la ponderación de la relación entre los beneficios y los riesgos y molestias previstos, y el consentimiento informado del paciente. Con respecto a sopesar la relación entre los beneficios esperados y los riesgos y molestias previstos, corresponde al análisis científico determinar, entre otras cuestiones, «cuánto» beneficio (directo o no directo) se espera obtener del estudio, «cuánto» riesgo y molestia se prevé para los pacientes que participarán en él y el margen de error de estas previsiones. A partir de esta información, al análisis ético le corresponde ponderar la relación entre estas variables y considerar si es moral realizar el estudio o no (esta cuestión la determinan, entre otros, los CEIC) y si vale la pena participar en él o no (decisión que toman los profesionales de la salud, los pacientes a los que se propone participar en el estudio clínico o sus familiares). La legislación vigente y las directrices internacionales señalan que un ensayo clínico sólo puede realizarse si los riesgos y molestias posibles se han sopesado y si no son desproporcionados en relación con los beneficios potenciales 26 que se puedan obtener11 y que corresponde al CEIC ponderar el balance de riesgos y beneficios anticipados que dimanan del estudio.12 Actualmente los protocolos de investigación y las hojas de información se limitan a señalar los beneficios que se espera obtener en el ensayo clínico y los riesgos y molestias que puede comportar, pero en ningún caso ponderan la relación entre éstos. Esta tarea deben realizarla los miembros del CEIC y los pacientes, tutores, curadores o guardadores de hecho. Cuando la proporción entre los beneficios y los riesgos y molestias es desproporcionada, el estudio clínico no obtiene el visto bueno del CEIC y por lo tanto no prospera. Pero si es proporcionada, el análisis y la determinación del valor de esta proporción, que suele tener un abanico muy amplio y a veces es de difícil determinación, recae en el paciente, tutor, curador o guardador de hecho. Sería recomendable que: i) Los promotores presentaran una valoración de la relación entre beneficios y riesgos y molestias a fin de que el CEIC la validara o corrigiera después de su propio análisis. Eso simplificaría la tarea de los CEIC y contribuiría a «promover una evaluación y una gestión apropiadas de los riesgos relacionados con la medicina, las ciencias de la vida y las tecnologías asociadas».13 ii) El paciente y su tutor, curador o guardador de hecho dispusieran de una orientación realizada por el investigador y supervisada por el CEIC, que situara el estudio clínico en una escala de valoración de la relación beneficios/riesgos-molestias justificada, que advirtiera del factor subjetivo que interviene en esta valoración (el riesgo y molestia que se considera que vale la pena asumir).14 El artículo 3.5 del Real decreto 223/2004 11 Consideración 4 y artículo 3.2.a de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo. Art. 3.c del RD 223/2004. Art. 14 de la Ley española 14/2007. Art. 18 de la Declaración de Helsinki (2008). 12 Art. 3.2.a de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo. Art. 12.2.c de la Ley española 14/2007 de investigación biomédica. 13 14 Art. 20 de la Declaración universal de bioética y derechos humanos (2005). En el momento de redactar este informe no tenemos noticia de la existencia de ninguna escala de ponderación beneficios/riesgos-molestias de los estudios clínicos; un instrumento que sería muy útil. 27 hace referencia a esto cuando dice que «tanto el umbral de riesgo como el grado de incomodidad deben ser definidos de forma específica y monitorizados durante el ensayo, especialmente cuando los sujetos del ensayo sean menores, adultos incapaces o constituyan una población especialmente vulnerable en razón de su situación económica, médica o social». 8 En la valoración de estudios clínicos hay que estar alerta a la maximización de los beneficios y la minimización de los riesgos que puede provocar el diferente ámbito de afectación (ciencia y futuros pacientes por un lado, paciente que participa en la investigación por el otro) 8.1 El ámbito de afectación. Entendemos por ámbito de afectación la persona o personas sobre las cuales se harán efectivos los posibles beneficios y los posibles perjuicios y molestias de un estudio clínico. El ámbito de afectación puede ser único o diferente. Es único cuando unas mismas personas obtienen u obtendrán los posibles beneficios y sufren los posibles riesgos o molestias del estudio clínico; y es diferente cuando unas personas obtienen los posibles beneficios y otras, los posibles riesgos y molestias. Los estudios con beneficios directos y no directos (BDyND)15 tienen un ámbito de afectación único y otro de diferente, porque los posibles beneficios, riesgos y molestias se harán efectivos en las personas que participan en el estudio (ámbito de afectación único), y también en futuros pacientes, que se beneficiarán del conocimiento obtenido y que no habrán sufrido los posibles riesgos y molestias y la incertidumbre propia de una investigación (ámbito de afectación diferente). En cambio, los estudios clínicos que sólo tienen beneficios no directos (BND) tienen un ámbito de afectación diferente, es decir, los posibles beneficios serán únicamente para los futuros pacientes, mientras que los posibles riesgos y molestias los sufren los pacientes que participan en la investigación. A la hora de valorar la relación entre los beneficios y los riesgos y molestias, los estudios clínicos con beneficios directos y no directos (BDyND) permiten hacer una comparación en un ámbito de 15 Los «estudios con beneficios directos (BD)» deben considerarse, de una manera estricta y tal y como se ha señalado en el punto 7.1, «estudios clínicos con posibles beneficios directos y no directos (BDyND)», porque cualquier estudio persigue validar hipótesis que beneficiarán a futuros pacientes. 28 afectación único, lo cual facilita medir «cuánto beneficio», «cuánto riesgo» y «cuánta molestia» se prevé en la persona afectada, ponderarlos y emitir un juicio, porque a pesar de la dificultad en medir y comparar diferentes parámetros, todos ellos hacen referencia y se concretan en unas mismas personas, lo cual disminuye el peligro de maximizar unos y minimizar otros (aunque, no hay que olvidarlo, este peligro continúa existiendo porque estos estudios también tienen un beneficio no directo y, por lo tanto, un ámbito de afectación diferente). Los estudios clínicos sólo con beneficios no directos (BND), en cambio, sólo tienen un ámbito de afectación diferente, lo cual dificulta medir y comparar los diferentes parámetros y aumenta el peligro de maximizar los beneficios y minimizar los riesgos y las molestias. Efectivamente, se corre el riesgo de multiplicar los beneficios por una n1 extraordinariamente alta (todas las personas afectadas presentes y futuras que se beneficiarán de los resultados de la investigación), mientras que los riesgos y las molestias se pueden multiplicar por una n2 baja («sólo» las personas que participarán en el ensayo clínico). En definitiva, puede pasar que, por un criterio utilitarista, se considere que los beneficios son muy superiores a los riesgos y molestias (BND > R-M), aunque los riesgos y las molestias sean muy elevados para las personas que participan en el estudio. Por este motivo hay que tener mucho cuidado con la interpretación de los artículos que establecen una proporcionalidad entre los beneficios y los riesgos y molestias sin especificar a qué tipo de beneficios se refieren; por ejemplo, el artículo 21 de la Declaración de Helsinki («La investigación médica en seres humanos sólo debe realizarse cuando la importancia de su objetivo es mayor que el riesgo inherente y los costes para la persona que participa en la investigación»), el 16 del Convenio de Oviedo («Que los riesgos en que pueda incurrir la persona no sean desproporcionados con respecto a los beneficios potenciales del experimento») o el 6.1 del Protocolo adicional al Convenio para la protección de los derechos humanos («La investigación no tiene que suponer para el ser humano un riesgo o restricción desproporcionado con respecto a los beneficios potenciales»). 29 8.2 El carácter absoluto y relativo de la ponderación. En el momento de valorar la relación entre los riesgos y molestias (R-M) y los beneficios no directos (BND), hay que considerar que, con personas que no tienen capacidad para decidir y en situaciones en las cuales no se esperan beneficios directos para el paciente que participa en el estudio clínico, los riesgos y molestias previstos tienen un valor absoluto, no proporcional. El carácter proporcional o relativo sólo lo puede otorgar la persona afectada cuando puede comparar el grado de riesgos y molestias que está dispuesto a asumir para contribuir a un bien. Este carácter absoluto lo encontramos en el artículo 27 de la Declaración de Helsinki, que, a pesar de lo que antes ha señalado en el artículo 21, dice que cuando se trata de personas incapaces el riesgo y el coste tienen que ser mínimos («Estas personas no deben ser incluidas en una investigación que no tenga posibilidades de beneficio para ellas, a menos que ésta tenga como objetivo promover la salud de la población representada por el individuo potencial, no pueda realizarse en personas competentes y la investigación implique sólo un riesgo y coste mínimos»). 9 Los beneficios, riesgos y molestias del estudio clínico deben compararse con los beneficios, riesgos y molestias del tratamiento convencional. Y si no hay tratamiento convencional, con los beneficios, riesgos y molestias que tendría el paciente si no recibiera ningún tratamiento y una correcta atención sanitaria 9.1 En la valoración de los estudios clínicos hay que perseguir el mínimo riesgo posible. Los beneficios, riesgos y molestias que se esperan de un estudio clínico deben compararse con los que tendría el paciente si se le administrara el tratamiento convencional, y si no hay tratamiento convencional, con los beneficios, riesgos y molestias que tendría sin recibir ningún tratamiento, pero con una correcta atención sanitaria.16 16 El artículo 32 de la Declaración de Helsinki habla de «la mejor intervención probada existente». Al respecto, la Introducción de las Pautas éticas internacionales para la investigación biomédica en seres humanos del Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS) en colaboración con la Organización Mundial de la Salud (OMS) (2002) dice lo siguiente: «El término utilizado comúnmente para describir el comparador activo éticamente preferible en ensayos clínicos controlados es el de "la mejor intervención existente". Sin embargo, en muchos casos hay más 30 Se ha señalado que un estándar de bajo riesgo es «cuando los riesgos de una intervención no son superiores a los asociados a un examen médico o psicológico de rutina».17 O que un riesgo es mínimo cuando «la probabilidad y la magnitud del daño y el malestar anticipados en el estudio no son por sí mismos mayores que los habitualmente observados en la vida cotidiana, mientras se realiza un ejercicio físico rutinario, en las exploraciones psicológicas o en los test».18 Consideramos que esta última definición de riesgo mínimo no es aceptable en la valoración de estudios clínicos, porque cuando hay tratamiento convencional éste reduce los riesgos con los cuales se encontraría la persona en la vida cotidiana sin tratamiento. Y cuándo no hay tratamiento convencional, porque una atención médica de acompañamiento, apoyo y cuidado en el proceso de enfermedad tiene efectos positivos en la salud del paciente. «Se considera que una investigación tiene un riesgo mínimo cuando, con respecto a la naturaleza y al alcance de la intervención, se puede esperar que comporte un impacto negativo muy débil y temporal sobre la salud de la persona afectada. Se considera que una investigación tiene una restricción mínima si se puede esperar que las contrariedades que pueden resultar sean temporales y muy ligeras para la persona afectada. Dado el caso, en el momento de la evade una intervención "existente" y los expertos clínicos no concuerdan en cuál es mejor. En otras circunstancias hay diversas intervenciones comprobadas "existentes" pero algunos expertos clínicos reconocen una como superior a las demás; otros prescriben una porque la mejor intervención no está, por ejemplo, disponible, o tiene un coste prohibitivo o es inapropiada para la capacidad de algunos pacientes de adherirse a un régimen completo y riguroso. El término usado en la pauta 11 para referirse a estas intervenciones, incluyendo la mejor alternativa y las posibles alternativas a la mejor, es "intervención de efectividad comprobada". En algunos casos, un comité de evaluación ética puede determinar que es éticamente aceptable utilizar una intervención de efectividad comprobada como elemento de comparación, incluso en casos en que esta intervención no es considerada como la mejor intervención vigente.» 17 Comentario sobre la pauta 9 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). 18 Hacen referencia a ello, por ejemplo, KARLAWISH, J. (2004): «Ethics of research in dementia» (traducción castellana: «Ética en la investigación sobre la demencia», en GAUTHIER, S [et al]: Enfermedad de Alzheimer y trastornos relacionados. Barcelona:Ars Medica, 2006, p. 155) y ARRIOLA MANCHOLA, E.; IBARZÁBAL ARAMBERRI, X. (2007): «Fundamentos éticos de respeto al enfermo», en MARTÍNEZ LAGE, J.M.; CARNERO PARDO, C. (editores): Alzheimer 2007: recapitulación y perspectivas, Madrid, Laboratorios Andrómaco, 2007, p. 283, que lo han extraído de: DEPARTMENT OF HEALTH AND HUMAN SERVICES (EE.UU.). «Common Rule, 45 CFR 46. Federal Policy for the Protection of Human Subjects; Notices and Rules», Federal Register, 1991, §102 (i). 31 luación de la restricción, se puede llamar a una persona de confianza de la persona afectada a fin de que evalúe la restricción.»19 10 El protocolo del estudio clínico tendría que diferenciar los beneficios y los riesgos y molestias directamente asociados al tratamiento experimental con posibles beneficios directos (R-MBD) de los beneficios y riesgos y molestias derivados de observaciones complementarias con beneficios no directos que persiguen aumentar el conocimiento científico (R-MBND), si los hay 10.1 Algunos estudios clínicos de los cuales se esperan beneficios directos incluyen intervenciones de control que no están directamente relacionadas con el tratamiento experimental. Son intervenciones para asegurar que el estudio proporciona conocimientos generalizables que responden a la pregunta de la investigación. Por lo tanto, a veces en un mismo estudio clínico puede haber: i) Riesgos y molestias asociados directamente al tratamiento. En este caso están asociados a posibles beneficios directos (R-MBD). ii) Riesgos y molestias asociados exclusivamente a las pruebas complementarias. En este caso están asociados a beneficios que no son directos para el paciente (R-MBND), aunque pueden ser necesarias para el estudio científico. Un ejemplo de riesgos y molestias asociados a pruebas complementarias son los ensayos en los cuales se prevén punciones lumbares para evaluar interacciones farmacológicas o metabólicas en el líquido cefalorraquídeo para conocer el funcionamiento del nuevo fármaco en el sistema nervioso central, sin que el facultativo que realiza la intervención con el paciente disponga de los resultados y, por lo tanto, sin que esta intervención influya en el tratamiento del usuario. En estas situaciones, dice Karlawish, «queda claro que el estudio consta de dos elementos distintos: un elemento son las intervenciones que constituyen tanto un riesgo como una posible ventaja (el nuevo fármaco), y otro elemento son las intervenciones que representan un riesgo pero que no aportan ninguna ventaja a los pacientes (la punción lumbar)». Y añade: «Para 19 Art. 17 del Protocolo adicional al Convenio para la protección de los derechos humanos y la dignidad del ser humano respecto de las aplicaciones de la biología y la medicina, relativo a la investigación biomédica (2005). 32 justificar de forma coherente los riesgos de un estudio deben analizarse por separado los riesgos de estos elementos. Los riesgos de los elementos con posibles ventajas sólo son justificables por sus posibles ventajas. Los riesgos de los elementos que no ofrecen ninguna ventaja están justificados por la importancia o el valor de la información que razonablemente pueda obtenerse con este estudio».20 Para facilitar la tarea de los CEIC, el protocolo del estudio clínico tendría que diferenciar y valorar por separado los beneficios y riesgos y molestias derivados del tratamiento experimental con posibles beneficios directos (R-MBD) de los beneficios y riesgos y molestias de las exploraciones complementarias que persiguen aumentar el conocimiento científico y que no tienen ningún beneficio directo para el paciente (R-MBND). 11 El CEIC debería evaluar y aprobar, por un lado, el tratamiento experimental y, por el otro y si las hay, las pruebas complementarias que persiguen aumentar el conocimiento científico 11.1 En estudios clínicos con riesgos y molestias derivados del tratamiento experimental con posibles beneficios directos (R-MBD) y con riesgos y molestias derivados de exploraciones complementarias que persiguen aumentar el conocimiento científico (R-MBND), el CEIC tendría que evaluar por separado el tratamiento experimental y las pruebas complementarias con el fin de poder aprobar uno y rechazar el otro, si llegara a considerarlo necesario. Pero actualmente, no es posible que los CEIC emitan dos dictámenes sobre un mismo protocolo de investigación. El RD 223/2004 no lo prevé y el Sistema Informático de Conexión de CEIC (SICCEIC) —que es la aplicación informática de gestión de los estudios clínicos— sólo permite un único dictamen por protocolo de investigación, que puede ser «favorable», «denegado» o «aclaraciones». Consideramos que habría que corregir este punto. La valoración del CEI debe tener en cuenta el papel que tiene la prueba complementaria para poder establecer un mecanismo de acción que pueda generar beneficios importantes en la sociedad 20 KARLAWISH, J. (2004): «Ethics of research in dementia» (traducción castellana: «Ética en la investigación sobre la demencia», o.c., p. 154-155). 33 (BND), como la curación o interrupción de la evolución de una enfermedad. 12 Los estudios clínicos dirigidos a personas incapaces de otorgar el consentimiento informado en los que los beneficios directos esperados son inferiores a los riesgos y molestias previstos (BD < R-M) o de los que no se prevén beneficios directos y sí riesgos y molestias (R-MBND), sólo deberían autorizarse cuando: (a) uno de los criterios de reclutamiento sea que el paciente haya autorizado, antes de la incapacidad y a través de un documento de instrucciones previas, participar en este tipo de estudios, o (b) el tipo de patología no haga posible promocionar las instrucciones previas en este tipo de investigación (p. ej., discapacidad intelectual congénita o traumatismo craneoencefálico). En esta última situación, la autorización debe ser muy prudente y exigente 12.1 El margen de libertad que otorgamos a las personas depende de tres factores. Las exigencias científicas respecto de los estudios dirigidos a personas que tienen plena capacidad de dar el consentimiento informado son o tendrían que ser las mismas que las de los destinados a personas que no la tienen. En cambio, las exigencias éticas y procedimentales no, dado que el margen de libertad que otorgamos a las personas para tomar decisiones varía en función de tres características: i) La capacidad o posibilidad de autogobernarse. Las personas con plena capacidad de autogobernarnos nos otorgamos un margen de libertad para aceptar incertidumbres, riesgos o molestias, y para rechazar beneficios, que vigilamos o que no damos a las personas que no la tienen, por ejemplo niños o personas con discapacidad intelectual. Por ello dispensamos una protección especial a las personas que no están en condiciones de dar su consentimiento a un ensayo clínico.21 Por ejemplo, consideramos que un adulto plenamente consciente es libre de participar en un estudio en fase I, pero no un niño o una persona con discapacidad intelectual. O encontraríamos incorrecto 21 La mayoría de las leyes y directrices señaladas en esta guía son un buen ejemplo de ello. Hacen mención especial a esta cuestión, por ejemplo, las consideraciones 3 y 4 de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo, y los artículos 7 y 8 de la Declaración universal sobre bioética y derechos humanos (2005). 34 que se aceptara la participación de una persona sin plena capacidad intelectual en un estudio clínico con riesgos y molestias sin haber hablado con las persones que los cuidan. ii) El ámbito de afectación de la decisión. El margen de libertad de una persona que toma una decisión que sólo le afecta a ella es mucho más amplio que el de una persona que decide por otra. Por otro lado, cuando hay que tomar decisiones que afectan a otras personas aumenta la exigencia de conocer y ponderar bien las consecuencias que tendrá la decisión antes de tomarla, porque la incertidumbre o la ligereza a la hora de tomar decisiones no es aceptable cuando afecta a otras personas. Hay decisiones, por ejemplo participar en una investigación clínica sin beneficios directos y con elevados riesgos y molestias, que sólo puede tomar la persona directamente afectada. iii) La cuestión por decidir. Actualmente, el margen de libertad para tomar una decisión no sólo depende de la capacidad de decidir, de la situación en la cual se encuentra la persona que tiene que tomar la decisión y del ámbito de afectación de la decisión, sino también del tipo de decisión. Hay decisiones, por ejemplo la eutanasia activa o el suicidio asistido, que se considera que la persona no puede tomar aunque tenga plena capacidad de autogobierno, la haya tomado serenamente y sólo le afecte directamente a ella. 12.2 Requisitos actuales para autorizar un estudio clínico sin beneficios directos con personas sin plena capacidad de decisión. Uno de los principales retos éticos de la investigación clínica con personas incapaces o que no tienen plena capacidad de otorgar el consentimiento es en qué condiciones es aceptable exponerlas a riesgos y molestias en un estudio del cual no se esperan beneficios directos. Los códigos deontológicos sanitarios y la legislación vigente establecen que las condiciones para hacer investigación con personas son las siguientes:22 22 Art. 16 del Convenio para la protección de los derechos humanos y la dignidad del ser humano respecto de las aplicaciones de la biología y la medicina (Convenio de Oviedo 1997/1999). Art. 9.5 de la Ley española 41/2002. Art. 20.1 de la Ley española 14/2007 de investigación biomédica. Art. 5. h de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo. Arts. 3, 5, 6, 15 y 17 del Protocolo 35 i) que los resultados de la investigación puedan producir beneficios reales o directos para la salud del paciente que participa en ella; ii) que los intereses del paciente siempre prevalecen sobre los intereses de la ciencia y de la sociedad; iii) que los riesgos a los cuales se expone la persona no sean desproporcionados en relación con los beneficios que se esperan de la investigación; iv) que sólo se puede realizar una intervención sanitaria en una persona incapaz de consentir si es a favor de su salud. Sin embargo, algunas declaraciones internacionales y la legislación señalan que se podrá autorizar una investigación que no comporte un beneficio directo para la salud de la persona si se cumplen las condiciones siguientes:23 i) que la investigación no se pueda llevar a cabo, con eficacia equiparable, con personas capaces de dar el consentimiento; ii) que la investigación tenga como objetivo, a través de una mejora significativa del conocimiento científico del estado de la persona, enfermedad o trastorno, contribuir a conseguir en un plazo determinado resultados que permitan obtener un beneficio para la persona afectada o para otras personas del mismo grupo de edad o que sufran la misma enfermedad o trastorno, o presenten las mismas características; iii) que los riesgos e inconvenientes a los cuales se expone la persona sean mínimos y proporcionados con relación a los beneficios que se esperan de la investigación, asumibles y admisibles; adicional al Convenio para la protección de los derechos humanos y la dignidad del ser humano con respecto a las aplicaciones de la biología y la medicina, relativo a la investigación biomédica (2005). Arts. 3 y 7 de la Declaración universal sobre bioética y derechos humanos (2005). Arts. 3, 4, 6, 17, 27 y 28 de la Declaración de Helsinki (2008). Art. 79 del Código de deontología del Consejo de Colegios de Médicos de Cataluña (2005). Arts. 3 y 7 de la Declaración universal sobre bioética y derechos humanos (2005). Arts. 3, 4, 6, 17, 27 y 28 de la Declaración de Helsinki (2008). 23 Arts. 6, 16 y 17 del Convenio de Oviedo (1997/1999). Arts. 3, 5 y 6 del RD 223/2004. Arts. 2. b y 20.2 de la Ley española 14/2007 de investigación biomédica. Norma 4.8.14 de la Guía de buena práctica clínica CPMP/ICH/95. Consideraciones 3 y 4 y artículos 3 y 5 de la Directiva 2001/20/CE. Pautas 9 y 13 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). Arts. 6.1 y 15 del Protocolo adicional al Convenio para la protección de los derechos humanos (2005). Art. 7 de la Declaración universal sobre bioética y derechos humanos (2005). 36 iv) que las intervenciones a que tienen que ser sometidos los sujetos del ensayo sean equiparables a las que corresponden a la práctica médica habitual en función de su situación médica, psicológica o social; v) que la persona no se oponga a ello o no se haya manifestado en contra previamente; vi) que la autorización de la investigación se ponga en conocimiento del ministerio fiscal.24 12.3 A las seis condiciones actualmente exigibles haría falta añadir la autorización en un documento de instrucciones previas, cuando haya sido posible obtenerlo. Consideramos que las actuales prescripciones legales para autorizar un estudio clínico con personas incapaces de otorgar el consentimiento informado y del cual no se prevén beneficios directos y sí riesgos y molestias, y por las razones que se exponen en la recomendación 12.4, no son suficientes para garantizar la dignidad de las personas que participan en él. Y que para garantizarla habría que añadir a las seis condiciones señaladas anteriormente (apartado 12.2) que uno de los criterios de reclutamiento sea que el paciente haya autorizado, antes de la incapacidad y a través de un documento de instrucciones previas, participar en este tipo de estudios, a menos que el tipo de patología lo haya hecho imposible. La posición 6 de la Position Statement Informed Consent for Research on Human Subjects with Dementia American Geriatrics Society Ethics Committee dice: «Los protocolos de investigación que no ofrezcan una expectativa razonable de beneficio directo a los sujetos que participan en ella, y que les exponga a un incremento de los riesgos mínimos, tendrían que ofrecerse sólo a los sujetos con capacidad para tomar decisiones o bien a los que lo han autorizado previamente. Debería establecerse un mecanismo nacional para examinar, caso por caso, los posibles proyectos de investigación que deben autorizarse, porque son muy prometedores, con sujetos que no tienen capacidad para tomar decisiones y no han manifestado voluntades anticipadas». 24 Esta condición la señala el artículo 20.2.c de la Ley española 14/2007 de investigación biomédica. 37 Somos conscientes de las repercusiones que puede tener en algunas líneas de investigación que las personas con una patología progresiva sólo puedan ser incluidas en estudios clínicos sin beneficios directos y con riesgos y molestias si lo han autorizado previamente. Para paliar las posibles repercusiones, la inclusión de esta nueva condición en nuestro ordenamiento jurídico (que el paciente haya autorizado previamente participar en este tipo de estudios, o que por el tipo de patología no haya sido posible redactar un documento de instrucciones previas) podría incluir una moratoria prudente de las condiciones actuales. Este hecho impulsaría y convertiría en una práctica habitual la redacción de documentos de instrucciones previas al inicio de las enfermedades de las que se espera una pérdida de la capacidad de decisión (véase la recomendación 39). Algunos estudios señalan que la mayoría de personas estudiadas no tendrían inconveniente en ser reclutadas para un estudio clínico sin beneficios directos y con molestias (p. ej., extracciones de sangre o punciones lumbares).25 Sin embargo, eso no diluye el problema ético del consentimiento por representación ni justifica no tener que disponer del consentimiento anticipado cuando ha habido posibilidad de obtenerlo. 12.4 Los argumentos que justifican esta recomendación son los siguientes: 12.4.1 El respeto a la dignidad de las personas es un precepto absoluto. No hay ningún derecho absoluto. La bioética suele recordar que los cuatro principios señalados por Beauchamp y Childress (autonomía, no maleficencia, beneficencia y justi- 25 KARLAWISH, J.; RUBRIGHT J.; CASARETT, D.; CARY, M.; TEN HAVE, T.; SANKAR, P. [«Older adults’ attitudes toward enrollment of non-competent subjects participating in Alzheimer’s research», American Journal of Psychiatry (febr. 2009), 166 (2), p. 182-188 (Epub 2008, 15 oct.)] entrevistaron a 538 personas mayores de 65 años para conocer su opinión sobre las personas con enfermedad de Alzheimer que participan en estudios clínicos, sobre el hecho de otorgar a un representante la potestad para autorizar la participación y sobre el margen de maniobra de éste para contradecir la voluntad del paciente. Los resultados fueron que la mayoría (83%) estaban dispuestos a otorgar consentimiento previo a estudios que comportaran una extracción de sangre, y casi la mitad (48%), a estudios con extracción de sangre y punción lumbar. La mayoría (96%) estaban dispuestos a nombrar un representante, y la mayoría, a concederle poderes de decisión (un 81% para la extracción de sangre y un 70% para la extracción de sangre y punción lumbar). 38 cia) son principios prima facie, es decir, normas que «en principio» hay que respetar pero que si entran en conflicto entre sí hay que determinar y justificar cuál de ellas prevalece. En este relativismo de la prevalencia de los diferentes principios deontológicos en diálogo entre sí y con las consecuencias, hemos establecido, sin embargo, un valor absoluto: la dignidad. Solemos considerar que todas las normas tienen excepciones, pero el respeto a la dignidad de la persona no las tiene. Este carácter inamovible y fundador ha sido reflejado en el artículo 10 de la Constitución española, qua otorga a la dignidad una categoría dogmática que ninguna ley puede alterar y sobre la cual se fundamenta el mismo orden político. En este sentido, el Tribunal Constitucional de España ha considerado que «la dignidad ha de permanecer inalterada cualquiera que sea la situación en que la persona se encuentre [...] constituyendo, en consecuencia, un “mínimum” invulnerable que todo estatuto jurídico debe asegurar».26 12.4.2 Que las personas tienen dignidad significa que tienen un valor en ellas mismas, que nunca pueden ser utilizadas simplemente como un medio. A pesar del carácter polisémico de la palabra dignidad,27 de la variedad de fundamentaciones que ha tenido y tiene,28 de la diversidad de aspectos que abraza según los contextos culturales y las personas que la reclaman, o incluso del hecho de que algunos autores lo juzgan un concepto inútil o incluso estúpido,29 consideramos necesario disponer de él para designar el valor que otorgamos a los seres humanos. Dignidad es el concepto que señala la «sacralidad» (que nosotros consideramos laica) que otorgamos a los seres humanos, que nos hace merecedores de un respe- 26 Sentencias del Tribunal constitucional español STC 120/1990 y STC 57/1994. 27 NORDENFELT, L. (2004): «The Varieties of Dignity», Health Care Analysis, 12 (2004), p. 69-81. 28 TORRALBA, F. (2005): ¿Qué es la dignidad humana? Ensayo sobre Peter Singer, Hugo Tristram Engelhardt y John Harris, Barcelona, Herder, 2005. 29 MACKLIN, R. (2003): «Dignity is a useless concept. It means no more than respect for persons or their autonomy», British Medical Journal, vol. 327, Londres, 2003, p. 1419-1420. PINKER, S. (2008): «The stupidity of dignity. Conservative bioethics’ latest, most dangerous ploy», The New Republic (28 de mayo de 2008). 39 to especial y que impide actos que podrían llegar a estar justificados por los principios o las consecuencias. En el caso que nos ocupa, hay ensayos clínicos que por más beneficios que comporten a la humanidad no están justificados si atentan contra la dignidad de la persona.30 El principio «el bienestar de la persona tiene que prevalecer sobre los intereses de la ciencia y de la sociedad» es heredero del imperativo kantiano sobre el cual se ha construido la moralidad moderna y la dignidad: «Obra de tal modo que uses a la humanidad, tanto en tu persona como en la persona de cualquier otro, siempre al mismo tiempo como fin y nunca simplemente como medio.»31 La dignidad, por lo tanto, está relacionada con aquello que tiene un valor en sí mismo y no puede ser sustituido por ninguna otra cosa ni puede ser utilizado simplemente como un fin. Se ha observado que el «simplemente» del imperativo kantiano («nunca simplemente como medio») es absolutamente necesario, porque muchas veces las personas actuamos o somos tratadas también como medio, por ejemplo cuando participamos voluntariamente en un estudio clínico, lo cual no afecta a nuestra dignidad. 12.4.3 La dignidad de las personas está relacionada con su autonomía y con la bondad de las acciones que esta autonomía les permite realizar. La dignidad de las personas está relacionada con su autonomía y con la bondad de las acciones que esta autonomía 30 En «Dignity is a useless concept», Ruth Macklin defiende que el concepto dignidad no aporta ningún otro significado o valor que no se exprese en estos cinco: (i) respeto a la persona, (ii) consentimiento informado, (iii) confidencialidad, (iv) evitar la discriminación y (v) evitar las prácticas abusivas. El concepto dignidad engloba, efectivamente, todos estos significados y seguramente más. Ahora bien, a Macklin le falta explicar por qué considera que debemos respetar a las personas, pedirles permiso, ser cuidadosos con sus datos y evitar la discriminación y las prácticas abusivas. Nosotros consideramos que lo tenemos que hacer precisamente porque les reconocemos la dignidad. 31 KANT, I. (1785): Grundlegung zur Metaphysik der Sitten, A66-A67 (trad. castellana de R. R. Aramayo: Fundamentación para una metafísica de las costumbres, Alianza Editorial, Madrid, 2002). En Metafísica de las costumbres lo expresa así: «La misma humanidad es una dignidad; porque el hombre no puede ser utilizado únicamente como medio por ningún hombre (ni por otros, ni tan solo por él mismo), sino siempre a la vez como un fin, y en esto consiste precisamente su dignidad» [KANT, I. (1797): Metaphysik der Sitten, II §38 (trad. castellana de A. Cortina y J. Conill: La metafísica de las costumbres. Segunda parte. Principios metafísicos de la doctrina de la virtud, Madrid, Tecnos, 2002)]. 40 les permite realizar. Tomás de Aquino lo expresó así: «He ahí el supremo grado de dignidad de los hombres: que por sí mismos, y no por otros, se dirijan hacia el bien».32 La autonomía y la bondad del acto permiten que una persona decida participar en un estudio clínico que no le comporta ningún beneficio y, por lo tanto, «convertirse en un medio», sin que eso suponga, ni mucho menos, que pierda la dignidad. Todo lo contrario: solemos considerarlo un acto muy digno, porque acepta riesgos y molestias sin ningún otro beneficio que ayudar a futuros enfermos. Es la dignidad propia de aquellos actos que, cuando traspasan un cierto umbral, consideramos heroicidades. En cambio, si se le obligase a participar en contra de su voluntad, o los motivos que le impulsaran a participar no fueran buenos (p. ej., porque lo hace únicamente para conseguir notoriedad o para hacer sufrir a alguien que le ama), lo consideraríamos indigno. Cuando una persona decide participar libremente y con capacidad para hacerlo en un estudio clínico sin beneficios directos, el «simplemente» del imperativo kantiano («nunca simplemente como medio») desaparece no porque a lo largo del estudio esta persona será tratada con respeto y dignidad en las formas, o porque ya se la trata como un fin en otros ámbitos, por ejemplo el familiar. Eso se podría dar y aun así su participación en el estudio clínico podría resultar indigna; por ejemplo, porque los fines del estudio o los motivos que le impulsan a participar en él no son buenos. El «simplemente» desaparece, y por lo tanto se mantiene la dignidad, porque ha decidido hacerlo libremente y porque la finalidad es buena, con lo cual en el «proceso en el que se convierte en medio» (el periodo de participación en el ensayo clínico) la persona no actúa simplemente como medio, sino también como una finalidad asumida por ella misma y que considera buena. Es, volvamos a repetirlo, la dignidad que otorgamos a los héroes y a los santos; y la indignidad de algunos actos que son realizados 32 TOMÁS DE AQUINO (1272?): Super Epistolas S. Pauli lectura. Ad Romanos, cap. II, lect. 3, n.º 217. 41 de manera obligada o persiguen fines perversos. La voluntad libre y consciente de la persona a participar en un estudio clínico que se considera bueno para la ciencia y la humanidad es, por lo tanto, primordial: señala la diferencia entre respetar su dignidad o la propia dignidad, o no hacerlo. Se suele decir que una lectura estricta del principio «el bienestar de la persona debe prevalecer sobre los intereses de la ciencia y de la sociedad» impediría cualquier investigación en la cual no haya beneficios directos para la persona que participa en ella;33 por ejemplo, todos los estudios en fase I,34 que por definición se realizan con voluntarios sanos. En este sentido, hay que destacar que la norma de prevalencia del bienestar de las personas sobre los intereses de la ciencia y la sociedad no es dogmática, sino un precepto prima facie. En el caso de estudios clínicos en fase I, se considera que la libertad de la persona (principio de autonomía) prevalece sobre el posible malestar que le pueda ocasionar (principios de beneficencia y no maleficencia en la tipología establecida por Beauchamp y Childress). 33 El Comité Consultatif National d’Éthique francés, en el Aviso n.º 73 sobre los ensayos de fase I en oncología (04/09/2002) señaló esta incoherencia de la Declaración de Helsinki (2000): « Los datos de la literatura científica muestran que en los ensayos en fase I en oncología —que siguen siendo indispensables— la relación beneficio/riesgo se inclina claramente del lado del riesgo. Estas pruebas están, por tanto, en contradicción con la Declaración de Helsinki, que las investigaciones clínicas deben atender. En la versión de octubre de 2000, esta declaración indica que "en la investigación médica en seres humanos, el bienestar de los sujetos siempre debe tener prioridad sobre los intereses de la ciencia y la sociedad". En el marco preciso de los ensayos en fase I en oncología, el término "bienestar de los sujetos" es abstracto y no aclara el alcance de la calidad de vida de los pacientes. Esta declaración considera que una investigación sólo estaría justificada si la población estudiada pudiera sacar un beneficio de ella. La distinción entre ensayos "con" y "sin beneficio directo" sólo existe en Francia. Y tendrá que ser suprimida después de la adaptación y publicación en Francia, antes del 1 de mayo de 2003, de las disposiciones necesarias para avenirse a la Directiva europea 2001/20/CE del 4 de abril de 2001. La supresión de la noción "sin beneficio individual directo" es una simplificación deseable pero podría llevar una deriva de los ensayos en fase I hacia riesgos para los pacientes. La redacción actual de esta directiva europea es ambigua y podría prohibir todos los ensayos de fase I en oncología, ya que el párrafo 2 del artículo 3 dice que "un ensayo clínico sólo se podrá iniciar si, en particular, (a) los riesgos e inconvenientes previsibles han sido sopesados en función del beneficio personal esperado por el sujeto que participa en el ensayo y para otros pacientes actuales o futuros" [...]». 34 En investigación clínica farmacéutica se diferencian cuatro etapas. La fase I se realiza con adultos sanos. La fase II, con un pequeño grupo de personas que sufren la enfermedad estudiada. La fase III, con un gran grupo de personas que sufren la enfermedad estudiada. Y la fase IV, también conocida como estudios de farmacovigilancia, se realiza después de que el producto ha conseguido permiso de comercialización y persigue recoger información sobre la seguridad del fármaco con un uso prolongado y la detección de toxicidad o reacciones adversas que no hayan sido detectadas anteriormente. 42 12.4.4 La donación voluntaria por solidaridad o amor es algo que solo puede decidir, salvo en casos excepcionales, la persona afectada. Se puede justificar la participación de una persona incapacitada en un ensayo clínico diciendo que, aunque no lo ha decidido libremente, la participación no atenta contra su dignidad porque los fines que se persiguen son buenos (el progreso de la medicina y el bienestar de pacientes futuros), y por lo tanto también lo son los motivos (la solidaridad y estimación de sus tutor para con los futuros pacientes). Pero la donación voluntaria por solidaridad o amor en un estudio clínico sin beneficios directos y con riesgos y molestias es algo que sólo puede decidir, salvo en casos excepcionales, la persona afectada. A las personas con una patología progresiva posible de diagnosticar antes de la pérdida de capacidad de decisión, ha habido la oportunidad de pedirles este acto de donación voluntaria. Y si no se hecho, esta negligencia u olvido no se debería paliar con la suplantación por parte del tutor de la autodonación voluntaria. S.Y.H. Kim y otros han realizado un estudio con una muestra representativa de norteamericanos mayores de 51 años a los que situaron en la hipótesis de no tener capacidad de decisión y en cuatro escenarios de investigación de diferentes grados de riesgo y de beneficio potencial: un ensayo clínico con punciones lumbares, uno de control de medicamentos, un estudio sobre vacunas o un estudio genético. Se les hicieron tres preguntas: (i) si se debe permitir el consentimiento por sustitución a la familia; (ii) si participarían en este tipo de ensayo, y (iii) si darían completa libertad de acción a su representante. La mayoría consideró que debe permitirse el consentimiento por sustitución en estos tipos de ensayo (del 67,5% al 82,5%, dependiendo del escenario), que ellos querrían participar en el ensayo (del 57,4% al 79,7%) y que darían una parte de libertad o libertad completa de decisión a sus representantes (del 54,8% al 66,8%), aunque este último resultado estaba relacionado con aquellos que estaban dispuestos a participar en el ensayo. El estudio detecta una tendencia a una disposición menor a 43 participar en este tipo de ensayos clínicos entre las personas pertenecientes a minorías étnicas y raciales.35 A pesar de estos resultados, o precisamente por estos resultados, hay que encontrar fórmulas para respetar la voluntad de las personas que no deseen o no deseaban participar en este tipo de estudios. 12.4.5 El argumento «él/ella hubiera querido participar en el ensayo» o «si él/ella pudiera, escogería participar», es un argumento lleno de dificultades que sólo se puede aceptar en casos excepcionales. El argumento de elegir lo que escogería el paciente si fuera capaz de hacerlo por sí mismo puede ser tan correcto como erróneo, y en la incertidumbre, lo más sensato es optar por la decisión más protectora. En casos excepcionales, este argumento puede ser válido siempre que se haga lo que se señala en la recomendación 33 sobre averiguar si la autorización se da en un contexto de cuidado y estima al paciente. En todo caso, la decisión siempre tiene que estar de acuerdo con los principios éticos y las creencias religiosas que el paciente practicó. 12.4.6 El peligro de las «patologías huérfanas» no justifica menospreciar la dignidad de las personas. También se suele recurrir al principio de igualdad y al hecho de que no realizar estudios de los que no se esperan beneficios directos para los pacientes que participan en ellos pero sí para el tratamiento conlleva el peligro de que algunas enfermedades se conviertan en «patologías huérfanas», con lo que las personas a las que se les diagnostican entrarían a formar parte de un grupo con dificultades para beneficiarse de la investigación clínica. Consideramos que este argumento sólo es válido para las patologías mentales que no se pueden prever o que son muy difíciles de prever, y siempre con las condiciones establecidas por la ley (véase el punto 12.2) y por esta guía. El desarrollo tecnocientífico del siglo XX ha hecho que des- 35 KIM, S.Y.H [et al.] (2009): «Surrogate consent for dementia research. A national survey of older Americans», Neurology, 2009, 72, p. 149-155. 44 confiemos de la relación directa que desde los griegos —y con algunos altibajos— habíamos establecido entre el bien, la verdad y el progreso científico. Nos hemos dado cuenta de que no todo lo que se puede hacer técnica y científicamente es bueno hacerlo y, en el caso que nos ocupa, que algunos estudios clínicos aumentarían enormemente el progreso biomédico y, por lo tanto, la lucha contra las enfermedades, pero que lo harían de un modo inmoral e indigno. 12.5 Tres supuestos sobre la posibilidad de poder redactar el documento de instrucciones previas o de no poder hacerlo. En lo referente a la posibilidad o no de redactar el documento de instrucciones previas, hay que diferenciar tres supuestos: i) Personas con una patología progresiva que es posible diagnosticar antes de la pérdida de capacidad de decisión (p. ej., la enfermedad de Alzheimer). Estas personas, que con una correcta información han podido prever la situación y dejar instrucciones en lo referente a su participación en estudios clínicos, sólo deberían poder ser incluidas en estudios clínicos sin beneficios directos y con riesgos y molestias si lo han autorizado previamente. ii) Personas con una pérdida brusca de la capacidad de decisión (p. ej., que han sufrido un traumatismo craneoencefálico) y que, por lo tanto, no necesariamente han podido prever su situación. Estas personas se regirían por los criterios actualmente vigentes. iii) Personas que presentan un grado variable de retraso mental y que nunca han tenido plena capacidad de decisión (p. ej., personas con síndrome de Down o síndrome X frágil). Estas personas se regirían por los criterios actualmente vigentes. 12.6 Cuando no ha sido posible obtener las instrucciones previas. Hay estudios clínicos que sólo se pueden hacer con personas con una patología que no ha hecho posible obtener las instrucciones previas —supuestos (ii) y (iii) del apartado anterior—; por ejemplo, personas con una discapacidad intelectual congénita o un traumatismo craneoencefálico. En estos casos no podemos recurrir al principio de autonomía que, como hemos dicho, en algunas situaciones señala la diferencia entre respetar la dignidad del pacien- 45 te o no hacerlo. Cuando ocurre esto y en estudios clínicos en los que los beneficios directos esperados son inferiores a los riesgos y molestias previstos (BD < R-M) o no se prevén beneficios directos y sí riesgos y molestias (R-MBND), es necesario hacer las apreciaciones siguientes: i) Son situaciones excepcionales y hay que velar para que lo sean. Se debe extremar, por lo tanto, el cumplimiento de las recomendaciones 1 y 2 con respecto al hecho de que la investigación clínica persiga, sin ningún tipo de dudas, beneficios para las personas que tienen la misma patología, que esté científicamente justificada y bien planteada y que sea imposible, con los conocimientos del momento, hacer el estudio con eficacia comparable con pacientes capaces de dar el consentimiento. ii) Hay que extremar también todas las demás recomendaciones de esta guía, especialmente aquellas que hacen referencia a la ponderación prudente de los riesgos, molestias, dolor, incomodidades, miedos, coerciones o cualquier perturbación del bienestar y la tranquilidad de la persona afectada. Sólo se pueden aceptar umbrales pequeñísimos de estos factores y nunca si la persona se ha manifestado o se manifiesta en contra de una manera u otra. iii) Es importantísimo que la autorización de los representantes legales se dé en una situación de cuidado y estima (véase la recomendación 33). iv) En la reunión del CEIC debería participar un representante de la asociación o asociaciones de afectados por la patología estudiada (recomendación 4). La autorización debería ser fruto de un consenso entre todos los miembros del CEIC y deberían extremarse las medidas de seguimiento del estudio (recomendación 41). 13 La inclusión en estudios clínicos de personas que se encuentran en una situación clínica de emergencia sin capacidad (temporal o permanente) de dar el consentimiento sólo se debería autorizar cuando, además de los requisitos señalados en la recomendación 12, haya probabilidades fundamentadas de que si participa en ellos obtendrá beneficios terapéuticos que no se pueden obtener de ningún otro modo. En el supuesto de estudios clínicos sin beneficios directos (BND), las condi- 46 ciones deberían ser las mismas que las señaladas en la recomendación 12 de esta guía 13.1 Nuestro ordenamiento jurídico y los protocolos de buenas prácticas. Nuestro ordenamiento jurídico y los protocolos de buenas prácticas dicen, con respecto a las situaciones de emergencia clínica, lo siguiente: 13.1.1 Artículo 21 de la Ley española 14/2007 de investigación biomédica: «1. Para la realización de una investigación en situaciones clínicas de emergencia, en las que la persona implicada no pueda prestar su consentimiento, deberán cumplirse las siguientes condiciones específicas: a) Que no sea posible realizar investigaciones de eficacia comparable en personas que no se encuentren en esa situación de emergencia. b) Que en el caso de que no sea previsible que la investigación vaya a producir resultados beneficiosos para la salud del paciente, tenga el propósito de contribuir a mejorar de forma significativa la comprensión de la enfermedad o condición del paciente, con el objetivo de beneficiar a otras personas con la misma enfermedad o condición, siempre que conlleve el mínimo riesgo e incomodidad para aquél. c) Que la autorización de la investigación se ponga en conocimiento del ministerio fiscal. »2. Se respetará cualquier objeción expresada previamente por el paciente que sea conocida por el médico responsable de su asistencia, por el investigador o por el Comité de Ética de la Investigación correspondiente al centro. »3. A los efectos del apartado primero de este artículo se consideran investigaciones en situaciones de emergencia, aquellas en las que la persona no se encuentre en condiciones de otorgar su consentimiento y, a causa de su estado y de la urgencia de la situación, sea imposible obtener a tiempo la autorización de los representantes legales del paciente o, de carecer de ellos, de las personas que convivieran con aquél. 47 »4. Las personas que participen en una investigación en situación de emergencia o, en su caso, sus representantes legales, deberán ser informados con la mayor brevedad posible en los términos establecidos en el artículo 4 de esta Ley. Asimismo se deberá solicitar el consentimiento para continuar participando en las investigaciones, en cuanto el paciente esté en condiciones de prestarlo.» 13.1.2 La norma 4.8.15 de la Guía de buena práctica clínica CPMP/ ICH/95 dice: «En situaciones de urgencia, cuando no sea posible obtener el consentimiento previo del sujeto, se solicitará el consentimiento del representante legal, si está presente. En los casos en que el consentimiento previo del sujeto no sea posible y el representante legal del sujeto no esté presente, la inclusión del sujeto deberá cumplir los requisitos descritos en el protocolo o en otra documentación aparte, con el dictamen favorable del CEIC para proteger los derechos, la seguridad y el bienestar del sujeto y asegurar el cumplimiento de los requisitos legislativos pertinentes. El sujeto o su representante legal será informado del ensayo en cuanto sea posible y deberá otorgar su consentimiento para continuar su participación en el mismo, si procede». 13.1.3 El comentario sobre la pauta 6 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002) prevé la posibilidad de proceder sin consentimiento informado en investigaciones de emergencia cuando (a) el paciente no pueda o no haya dado el consentimiento; (b) no se dispone de tiempo suficiente para localizar a una persona que pueda autorizar la participación, y (c) no sea posible identificar previamente la población que probablemente pueda sufrir la condición estudiada. 13.2 La normativa vigente no garantiza suficientemente la dignidad y los derechos de los posibles participantes. Consideramos que el artículo 21.1.b de la Ley española 14/2007 de investigación bioética, la norma 4.8.15 de la Guía de buena práctica clínica CPMP/ICH/95 y el comentario sobre la pauta 6 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002) no garantizan suficientemente la dignidad y los derechos de los posibles participantes, por las razones siguientes: 48 i) Hay que especificar que la inclusión de un paciente en un estudio clínico en una situación de urgencia clínica y sin que sea posible obtener su consentimiento requiere que haya probabilidades fundamentadas de que obtendrá beneficios terapéuticos que no se pueden obtener de ninguna otra manera. ii) Que los estudios clínicos de los cuales no se esperan beneficios directos para los pacientes que participan y sí para otras personas con la misma enfermedad o condición sólo deberían autorizarse en las condiciones señaladas en la recomendación 12 (que uno de los criterios de reclutamiento sea que el paciente haya autorizado previamente su participación en este tipo de estudios, a menos que el tipo de patología lo haga imposible). III. ESTUDIOS CON PLACEBO 14 En investigaciones clínicas con personas adultas incapaces o que no tienen plena capacidad de otorgar el consentimiento informado, sólo es aceptable que el grupo control tome placebo o no reciba ningún tratamiento en estudios para los cuales no hay tratamiento convencional de eficacia probada o, en caso de que lo haya, no tiene eficacia en los posibles participantes en el estudio 14.1 La normativa vigente. La legislación y el Código deontológico del Consejo de Colegios de Médicos de Cataluña señalan que no se puede negar o interrumpir una terapia eficaz reconocida para ensayar nuevos tratamientos a menos que, después de una esmerada información, el enfermo dé su consentimiento expreso. Concretamente, el artículo 24 de la Ley española 14/2007 de investigación biomédica dice: «(1) La investigación no deberá retrasar o privar a los participantes de los procedimientos médicos preventivos, diagnósticos o terapéuticos que sean necesarios para su estado de salud. (2) En el caso de investigaciones asociadas con la prevención, diagnóstico o tratamiento de enfermedades, deberá asegurarse que los participantes que se asignen a los grupos de control reciban procedimientos probados de prevención, diagnóstico o tratamiento. [...] (3) Podrá recurrirse al uso de placebo sólo si no existen métodos de eficacia probada, o cuando la retirada de estos métodos no presente un riesgo o perjuicio inaceptable para el paciente». 49 El artículo 32 de la Declaración de Helsinki (2008) dice: «Los posibles beneficios, riesgos, costes y eficacia de toda intervención nueva tienen que ser evaluados mediante la comparación con la mejor intervención probada existente, excepto en las circunstancias siguientes: (a) El uso de un placebo, o ningún tratamiento, es aceptable en estudios para los cuales no hay ninguna intervención probada existente. (b) Cuando por razones metodológicas, científicas y urgentes, el uso de un placebo es necesario para determinar la eficacia y la seguridad de una intervención que no implique un riesgo, efectos adversos graves o daño irreversible para los pacientes que reciben el placebo o ningún tratamiento. Hay que tener mucho cuidado para evitar abusar de esta opción». La pauta 11 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002) dice: «Por regla general, los sujetos de investigación en el grupo control de un ensayo de diagnóstico, terapia o prevención, deberían recibir una intervención de efectividad comprobada. En algunas circunstancias, puede ser éticamente aceptable utilizar un control alternativo, como placebo o ausencia de tratamiento. El placebo se puede usar: (a) cuando no exista una intervención de efectividad comprobada; (b) cuando la omisión de una intervención de efectividad comprobada expondría a los sujetos, a lo sumo, a una molestia temporal o a un retraso en el alivio de los síntomas; (c) cuando el uso de una intervención de efectividad comprobada como control no produciría resultados científicamente de confianza y el uso de placebo no añadiría ningún riesgo de daño serio o irreversible para los sujetos». 14.2 Razones para la utilización de placebo. Se suele justificar el uso del placebo en estudios clínicos a pesar de haber tratamiento convencional:36 36 LEVINEM, R.J. (1999): «The need to revise the Declaration of Helsinki», New England Journal of Medicine, 341, p. 531-534. TEMPLE, R.; ELLENBERG, S.S. (2000): «Placebo controlled trials and active control trials in the evaluation of new treatment. I: ethical and scientific issues», Annals of Internal Medicine, 133, p. 455-463. ELLEBERG, S.S.; TEMPLE, R. (2000): «Placebo controlled trials and active control trials in the evaluation of new treatment. II: practical issues and specific cases», Annals of Internal Medicine, 133, p. 464-470. LYONS, D.J. (1999): «Use and abuse of placebo in clinical trials», Drug Information Journal, 33, p. 261-264. 50 i) Por criterios de eficiencia, porque los estudios con placebo son más baratos y rápidos (necesitan una muestra más reducida) que aquellos que se comparan con el fármaco convencional (diseño head to head), lo cual permite que los productos estudiados puedan ser evaluados y aprobados más rápidamente y la población enferma que los necesita se pueda beneficiar de ellos. ii) Por criterios metodológicos, porque comparar dos fármacos puede mostrar que uno es mejor que el otro, pero si no se compara con placebo podría ser que ninguno de los dos fuera bastante efectivo para demostrar diferencias significativas. iii) Cuando el tratamiento convencional no mejora la salud del paciente ni la calidad de vida de su entorno. iv) Cuando el tratamiento experimental no persigue tener más eficacia que el convencional, sino disponer de una gama terapéutica más amplia para una misma patología con el fin de poder hacer frente a las diferentes idiosincrasias de los pacientes, que a veces no toleran bien el tratamiento convencional (medicamentos de la misma clase que otro ya reconocido pero que necesitan pasar por un ensayo clínico para conseguir ser autorizados. Se suelen llamar «medicamentos me-too»). En general, este tipo de argumentación se puede considerar poco científica, porque aquello que interesa saber del nuevo tratamiento es si es superior, igual o inferior al convencional en eficacia, seguridad, gasto, comodidad o tolerabilidad. Y eso sólo se consigue con un ensayo head to head.37 v) Por respeto a la libertad de las personas, que pueden decidir participar en un estudio clínico asumiendo algunos riesgos. vi) Porque se trata de afecciones leves (p. ej., acné) o con posibles consecuencias negativas pequeñas (p. ej., casos de depresión no grave) y en cambio el diseño con dos tratamientos activos (head to head) sería muy complejo y costoso. 14.3 Supuestos en los cuales el grupo control puede tomar placebo o no recibir ningún tratamiento. Sin entrar en la valoración de la 37 BOFILL, X.; URRUTIA, G.; ALONSO, P.; ROURA, M. (2007): «La participación de los pacientes en los ensayos clínicos», Humanitas. Humanidades Médicas, nº 17, julio 2007. 51 apreciación del artículo 24.3 de la Ley española 14/2007 («cuando la retirada de estos métodos no presente un riesgo o perjuicio inaceptable para el paciente»), ni del apartado b del artículo 32 de la Declaración de Helsinki («cuando la omisión de una intervención de efectividad comprobada expondría a los sujetos, a lo sumo, a una molestia temporal o a un retraso en el alivio de los síntomas»), que entendemos que se refieren a personas con capacidad para otorgar el consentimiento informado, consideramos que en personas adultas incapaces o que no tienen plena capacidad de otorgar el consentimiento, el grupo control sólo puede tomar placebo o no recibir tratamiento en alguno de estos supuestos y siempre que los beneficios esperados sean superiores a los riesgos y molestias: i) en estudios clínicos para los cuales no hay tratamiento convencional de eficacia probada; ii) en estudios clínicos para los cuales el tratamiento convencional no tiene eficacia en las personas susceptibles de participar en el estudio clínico; iii) el paciente ha autorizado en un documento de instrucciones previas su participación en este tipo de estudio. IV. CAPACIDAD DEL PACIENTE PARA TOMAR DECISIONES 15 Evaluar la competencia de una persona para dar el consentimiento es una tarea de mucha responsabilidad profesional y moral. Hay que evaluar cada caso concreto utilizando procedimientos reconocidos, la pericia profesional y, cuando haga falta, la decisión participada 15.1 No hay una necesaria relación entre trastorno mental e incapacidad para la toma de decisiones. En primer lugar es necesario advertir que evaluar la competencia de una persona para consentir o rechazar participar en un estudio clínico no es tarea del CEIC, sino del médico responsable del reclutamiento.38 38 En este sentido, el artículo 3a de la Ley española 41/2002 dice que son situaciones de otorgamiento del consentimiento por representación «cuando el paciente no sea capaz de tomar decisiones, a criterio del médico responsable de la asistencia […]». 52 Los profesionales sanitarios tienen criterios para diagnosticar los trastornos mentales e instrumentos para medir la gravedad de la sintomatología. No cabe duda de que la gravedad de algunas patologías mantiene una clara concordancia con la capacidad para tomar decisiones; por ejemplo, una enfermedad de Alzheimer grave (GDS-FAST ≥ 6), un retraso mental profundo (CI < 20) o una sintomatología psicótica grave. Ahora bien, la capacidad de decisión no se muestra siempre como un rasgo dicotómico e inalterable, en el sentido que uno sea o no capaz de tomar decisiones y por siempre y en todo momento, sino como un estado que depende de una variedad de factores funcionales en un continuo que puede variar en el tiempo y según el contexto y las circunstancias. La decisión de una persona se puede ver afectada: i) por las características de la persona (cognición y voluntad); ii) por el estado en que se encuentra esta persona en el momento en que tiene que tomar la decisión (estado emocional); iii) por la situación en que se encuentra esta persona en el momento en que debe tomar la decisión (situación social); iv) por el riesgo y la dificultad de la decisión y las alternativas posibles (decisión a tomar), y v) por la calidad del apoyo y el acompañamiento que recibe en el proceso de información y decisión (calidad de los procedimientos de autorización informada). Las dos primeras características se centran en la capacidad del sujeto, mientras que las otras tres lo hacen en la relación que mantiene con el entorno. Como veremos más adelante, se han creado escalas para valorar la capacidad para tomar decisiones, y algunos estudios indican que no hay una necesaria correlación entre estos valores y los obtenidos en las escalas de medición del trastorno mental,39 lo cual señala también que no hay una necesaria relación entre 39 Hay que destacar, entre otros: DILIP, V.; JESTE, M.D.; SAKS, E.S. (2006): «Decisional capacity in mental illness and substance use disorders: empirical database and policy implications», Behavioral Sciences and the Law, 24, p. 607-628. BARTON, W.; PALMER, PH.D. [et al.] (2005): «Assessment of capacity to consent to research among older person with schizophrenia, Alzheimer disease, or diabetes mellitus», Archives of General Psychiatry, vol. 62, julio 2005. VICTOR, W.C. LUI [et al.] (2009): «Capacity to make treatment decisions in Chinese older persons with very mild dementia and mild Alzheimer disease», American Journal of Geriatric Psychiatry, 17 (5), p. 428-436. SIMÓN-LORDA, P. (2008): «La capacidad de los pacientes para tomar decisiones: una tarea todavía pendiente», o.c. 53 trastorno mental e incapacidad para la toma de decisiones, y que es aconsejable un examen de capacidad de decisión individualizado.40 Jurídicamente, la capacidad para tomar la decisión de participar o no en un estudio clínico es categórica: una persona es competente o no, es capaz o no. En ética y en la práctica clínica, en cambio, la capacidad de decisión es gradual, y el abanico de posibilidades que se despliega sólo puede ser acotado valorando cada situación concreta. Un diagnóstico de demencia, por ejemplo y tal como señala la posición segunda del American Geriatrics Society Ethics Committee, no confiere automáticamente la incapacidad en la toma de decisiones. Especialmente en las primeras etapas de la demencia, muchas personas continúan siendo capaces de tomar decisiones, incluida la de decidir si participan o no en una investigación.41 15.2 Instrumentos para medir la capacidad de decisión. Las características del ejercicio de la autonomía personal han ocupado sobradamente a filósofos y estudiosos en general, pero no ha sido hasta la aparición de la bioética y del reconocimiento efectivo de la autonomía del paciente cuando ha sido necesario disponer de parámetros objetivos para determinar la capacidad de una persona para tomar decisiones clínicas. Sin embargo, aunque la evaluación de la capacidad de decisión es un aspecto primordial en el proceso de consentimiento informado y de peritaje, es una cuestión todavía no resuelta satisfactoriamente. En 1977, Roth, Meisel y Lidz publicaron lo que se suele considerar el primer artículo explícitamente dedicado a la evaluación de la capacidad de decisión. En este trabajo se revisaban los criterios utilizados en la práctica habitual del mundo clínico y judicial, se proponía un test con cinco criterios para evaluar y se abordaba la relación entre el «balance de los beneficios y riesgos» (que podía ser 40 El estudio de WARNER, J.; MCCARNEY, R.; GRIFFIN, M.; HILL, K.; FISHER, P. [«Participation in dementia research: rates and correlates of capacity to give informed consent», Journal of Medical Ethics (marzo 2008), 34 (3), p. 167-170], por ejemplo, indica que las pruebas cognitivas son insuficientes para evaluar la capacidad de decisión. Sobre la capacidad para tomar decisiones, véase BOADA ROVIRA, M.; ROBLES BAYÓN, A. (ed.) (2009): Documento Sitges 2009. Capacidad para tomar decisiones durante la evolución de una demencia: reflexiones, derechos y propuestas de evaluación, Barcelona, Editorial Glosa, 2009. 41 AMERICAN GERIATRICS SOCIETY ETHICS COMMITTEE (2007): Position Statement Informed Consent for Research on Human Subjects with Dementia AGS Ethics Committee. 54 favorable o desfavorable) y si la decisión era de consentimiento o de rechazo al tratamiento. Sobre esta última cuestión, los autores consideraban que es necesaria una baja capacidad psíquica para aceptar un tratamiento favorable y para rechazar uno desfavorable y, en cambio, una capacidad alta para rechazar un tratamiento favorable y aceptar uno desfavorable.42 Unos años más tarde, esta cuestión fue retomada por la Escala Móvil de Capacidad. Actualmente disponemos de diversos protocolos para evaluar la capacidad de consentimiento43, que se agrupan en dos grandes bloques: (i) instrumentos para evaluar la capacidad de dar el consentimiento para recibir un tratamiento y (ii) instrumentos para evaluar la capacidad de dar el consentimiento para participar en un estudio clínico. Ninguno de estos dos bloques dispone actualmente del reconocimiento unánime de la comunidad científica ni, en el momento de redactar estas líneas, hay ninguno validado en lengua castellana. i) El instrumento más utilizado para evaluar la capacidad de dar el consentimiento para recibir un tratamiento es el MacArthur Competence Assessment Tool-Treatment (McCAT-T).44 Requie42 ROTH, L.H.; MEISEL, A.; LIDZ, C. W. (1977): «Test of Competency to Consent to Treatment», American Journal of Psychiatry, 1977, 134 (3), p. 279-286. 43 Para una revisión de la defensa y críticas de los diferentes protocolos de evaluación de la capacidad, véase STURMAN, E.D. (2005): «The capacity to consent to treatment and research: a review of standardized assessment tools», Clinical Psycology Review, 2005, 25, p. 954-974. MOYE, J. [et al.] (2006): «Empirical advances in the assessment of the capacity to consent to medical treatment. Clinical implications and research needs», Clinical Psycology Review, 2006, 26, p. 1054-1077. 44 APPELBAUM, T.L.; GRISSO T. (1986): A history and theory of informed consent, New York, Oxford University Press, p. 274-297. (1988): «Assessing patient’s capacities to consent to treatment», New England Journal of Medicine (NEJM), 1988, 319 (25), p. 1635-8. (1995): «The MacArthur treatment competence study», Law and Human Behavior, 19, p. 105-174. (1998): Assessing competence to consent to treatment: a guide for physicians and other health professionals, New York, Oxford University Press. (2001): MacCAT-CR: MacArthur Competence Assessment Tool for Clinical Research, Sarasota (Florida), Professional Resource Press. También hay la MacArthur Competence Assessment Tool-Criminal Adjudication (McCAT-CA). KARLAWISH ha resumido y adaptado las preguntas de la escala MacArthur Competence Assessment Tool for Clinical Research para evaluar la capacidad de saber entender, apreciar, razonar y elegir en «Ethics of research in dementia» (traducción castellana: «Ética en la investigación sobre la demencia», en GAUTHIER, S. [et al.]: Enfermedad de Alzheimer y trastornos relacionados, Barcelona, Ars Medica, 2006, p. 147). En cuanto a la contrastación empírica de la MacArthur Competence Assessment Tool for Clinical Research, véase KARLAWISH J.; KIM S.Y.; KNOPMAN, D.; VAN DYCK, C.H.; JAMES, B.D.; MARSON, D. (2008): «Interpreting the clinical significance of capacity scores for informed consent in Alzheimer disease clinical trials», American Journal of Geriatric Psychiatry (julio 2008), 16 (7), p. 568-574 (Epub, 12 junio 2008). 55 re una entrevista en la cual se valoran cuatro apartados: (1) «entender» (capacidad de comprender el significado de la información enviada); (2) «apreciar» (capacidad de identificar la manera de aplicar la información recibida a la propia situación personal); (3) «razonar» (capacidad de comparar opciones —razonamiento comparativo— y de deducir las consecuencias de estas opciones — razonamiento consecutivo—), y (4) «elegir» (capacidad de dar a conocer de manera coherente una decisión). Algunos estudios comparativos consideran que otras escalas pueden ser iguales o mejores en determinadas situaciones.45 De estas otras escalas cabe destacar: Aid to Capacity Evaluation (ACE),46 Capacity Assessment Tool (CAT),47 Competency to Consent to Treatment Instrument (CCTI),48 Decision Assessment Measure (DAM),49 Hopemont Capacity Assessment Interview (HCAI),50 Competency Interview Schedule (CIS),51 Ontorio Competency Questionnaire (OCQ),52 Fitten's Clinical Vignettes (FCV),53 Hopkins Competency Assessment Tool (HCAT)54 y Structured Interview 45 DUNN, L. B. [et al.] (2006): «Assessing decisional capacity for clinical research or treatment: A review of instruments». American Journal of Psychiatry (agosto 2006), 163, p. 1323-1334. 46 ETCHELLS, E. [et al.] (1999): «Assessment of patients capacity to consent to treatment», Journal of General Internal Medicine, 1999, 14, p. 27-34. 47 CARNEY, M. [et al.] (2001): «The development and piloting of a capacity assessment tool», The Journal of Clinical Ethics, 2001, 12, p. 17-23. 48 MARSON MCCODY [et al.] (1995): «Neuropsychological predictors of competency in Alzheimer's disease using a rational reason legal standard», Archives of Neurology, 1995, 52, p. 955-959. 49 WONG, J.G. [et al.] (2000): «The capacity of people with a mental disability to make a health care decision», Psychological Medicine, 2000, 30, p. 295-306. 50 EDELSTEIN, B. (1999): Hopemont capacity assessment interview manual and scoring guide, Mongantown, W.V., West Virginia University, 1999. 51 BEAN, G. [et al.] (1994): «The psychometric properties of the competency interview schedule», Canadian Journal of Psychiatry, 39, p. 368-376. 52 DRAPER, R.J.; DAWSON, D. (1990): «Competence to consent to treatment:A guide for the psychiatrist», Canadian Journal of Psychiatry, 35, p. 285-289. 53 FITTEN, L.J.; WAITE, M.S. (1990): «Impact of medical hospitalization on treatment decisionmaking capacity in the elderly», Archives of Internal Medicine, 150, p. 1717-1721. 54 JANOFSKY, J.S.; MCCARTHY, R.J.; FOLSTEIN, M.F. (1992): «The Hopkins Competency Assessment Test: A brief method for evaluating patients' capacity to give informed consent», Hospital and Community Psychiatry, 43, p. 132-136. 56 for Competency and Incompetency Assessment and Ranking Investory (SICIATRI).55 Es interesante la aportación de la Escala Móvil de Capacidad defendida y desarrollada por James Drane56 y Allen Buchanan y Dan Brock.57 En esta escala se considera que el umbral por debajo del cual la persona es incapaz de tomar una decisión es variable y depende de la importancia y gravedad de la decisión. Una persona puede tener capacidad para dar el consentimiento a tratamientos o estudios clínicos de bajo riesgo y mucho beneficio, pero no de alto riesgo y complicados. Esta escala móvil se divide en tres niveles que dependen del balance beneficio/riesgo, de tal manera que se exige más capacidad para tomar decisiones cuanto mayor es el riesgo y mayor el posible mal, y en estos casos, también se exige más certeza en la valoración. De entre las críticas que ha recibido la Escala Móvil de Capacidad hay que destacar las de Wilks, Cale y Demarco58, Wicclair59 y Beauchamp y Childress.60 ii) Los instrumentos más utilizados para evaluar la capacidad de dar el consentimiento para participar en un estudio clínico son los siguientes: 55 TOMODA, A. [et al.] (1997): «Validity and reliability of structured interview for competency incompetency assessment testing and ranking inventory», Journal of Clinical Psychology, 53, p. 443-450. 56 DRANE, J. (1984): «Competency to give an informed consent. A model for making clinical assessments», JAMA (The Journal of the American Medical Association), vol. 252, n.º 7, p. 925-927. (1985): «The many faces of competency», The Hastings Center Report, vol. 15, n.º 2, p. 17-21. 57 BUCHANAN, A.; BROCK, D.W. (1989): Deciding for others. The ethics of surrogate decisionmaking, New York, Oxford University Press. BUCHANAN, A. (2004): «Mental capacity, legal competence and consent to treatment», Journal of de Royal Society of Medicine, n.º 97, p. 415-420. 58 WILKS, I. (1997): «The debate over risk-related standards of competence», Bioethics, 11, p. 419420. (1999): «Asymmetrical competence» Bioethics, 13, p. 154-159. CALE, GR. S. (1999): «Riskrelated standards of competence: Continuing the debate over risk-related standards of competence», Bioethics, 13, p. 131-148. DEMARCO, J.P. (2002): «Competence and paternalism», Bioethics, 16 (3), p. 231-245. 59 WICCLAIR, M.R. (1991): «Patient decision-making capacity and risk», Bioethics, vol. 5, n.º 2, p. 95. (1991): «A response to brock and skene», Bioethics, vol. 5, n.º 2, p. 120. 60 BEAUCHAMP, T.L.; CHILDRESS, J.F. (1994): Principles of biomedical ethics (Fourth Edition) (trad. castellana de Gracia, T., Júdez F.J. y Feito, L.: Principios de ética biomédica, Masson, Barcelona 1998, p. 130-134). 57 • MacArthur Competence Assessment Tool-Clinical Research (McCAT-CR).61 El más utilizado y reconocido para evaluar la capacidad de dar el consentimiento informado en un estudio clínico. El tiempo de administración es de 15 a 30 minutos, y el formato, una entrevista semiestructurada. • Evaluation to Sign Consent (ESC).62 El tiempo de administración es de 5 a 10 minutos y el formato es un cuestionario de 5 temas después de haber realizado el proceso de información. • Quality of Informed Consent (QuIC).63 El tiempo de administración es inferior a 10 minutos y tiene una parte objetiva de diversas opciones y una parte subjetiva de comprensión. • Deaconess Informed Consent Comprehension Test (DICCT).64 El tiempo de administración es inferior a 10 minutos, y el formato, una entrevista estructurada. • Two-part Consent Form.65 Una parte autoadministrada y una entrevista semiestructurada. • California Scale of Appreciation (CSA).66 El tiempo de administración es de 10 a 20 minutos y el formato es una entrevista estructurada. • Competency Assessment Interview (CAI).67 Entrevista estructurada, con viñetas. 61 APPELBAUM, T.L.; GRISSO T. (2001): MacArthur Competence Assessment Tool for Clinical Research, Sarasota (Florida), Profesional Resource Press. 62 DERENZO, E.G.; CONLEY, R.R; LOVE, R.C. (1998): «Assessment of capacity to gide consent to research participation: State of the art and beyond», The Journal of Health Care Law and Policy, 1, p. 66-87. 63 JOFFE, S. [et al.] (2001): «Quality of informed consent: A new measure of understanding among research subjects», Journal of the National Cancer Institute, 93, p. 139-147. 64 MILLER, C.K. [et al.] (1996): «The Deaconess Informed Consent Comprehension Test: An assessment tool for clinical research subjects», Pharmacotherapy, 16, p. 872-878. 65 ROTH, L.H. [et al.] (1982): «Competency to decide about treatment or research: An overview of some empirical data», International Journal of Law and Psychiatry, 5, p. 29-50. MILLER, R.; WILLNER, H.S. (1974): «The Two-part Consent Form: a suggestion for promoting free and informed consent», New England Journal of Medicine, 290, p. 964-966. 66 SAKS, E.R. (2002): «The California Scale of Appreciation: A new instrument to measure the appreciation component of capacity to consent to research», American Journal of Geriatric Psychiatry, 10, p. 166-174. 67 STANLEY, B. [et al.] (1984): «The elderly patient and informed consent: empirical findings», JAMA, 252, p. 1302-1306. 58 • Informed Consent Survey (ICS)68. El tiempo de administración es de unos 15 minutos, y el formato, una entrevista estructurada. • Brief Informed Consent Test (BICT).69 El tiempo de administración es de 5 a 10 minutos y el formato es un cuestionario de 11 ítems a los que hay que responder sí o no. • Vignette methods described by Schmand et al.70 y Sanchs et al.71 El tiempo de administración es de unos 15 minutos y el formato es una entrevista estructurada. • University of California, San Diego Brief Assessment of Capacity to Consent (UBACC).72 15.3 Experiencia del profesional y decisión participada. Como se ha señalado anteriormente, ningún instrumento para medir la capacidad de decisión goza del acuerdo unánime de la comunidad científica. Hay que considerar, además, que la incapacidad de una persona para el consentimiento no es un parámetro que se pueda determinar únicamente con un cuestionario o entrevista de 10 o 20 minutos, y que siempre es necesario disponer del historial clínico y evaluarlo, y tener en cuenta el contexto en el cual los agentes toman la decisión. Por otra parte, en cualquier escala valorativa hay una zona intermedia de incertidumbre o penumbra en la cual se hace más difícil determinar si la persona es capaz de tomar una decisión o no. Por este motivo, además de los procedimientos reconocidos, es necesario recurrir a la pericia profesional (que considerará las variables que aparecen en el historial clínico) y, cuando sea necesario, a la decisión participada (véase la recomendación 35). 68 WIRSHING, D.A. [et al.] (1998): «Informed consent: Assessment of comprehension», American Journal of Psychiatry, 155, p. 1508-1511. 69 BUCKLES, V.D. [et al.] (2003): «Understanding of informed consent by demented individual», Neurology, 61, p. 1662-1666. 70 SCHMAND, B. [et al.] (1999): «Assessment of mental competency in community-dwelling elderly», Alzheimer Disease and Associated Disorders, 13, p. 80-87. 71 SACHS, G. [et al.] (1994): «Ethical aspects of dementia research: informed consent and proxy consent», Journal of Clinical Research, 42, p. 403-412. 72 DILIP, V.; JESTE, M.D. [et al.] (2007): «A new brief instrument for assessing decisional capacity for clinical research», Archives of General Psychiatry, 64 (8), p. 966-974. 59 15.4 La autonomía tiene una vertiente social de reconocimiento. Para ejercer la autonomía a veces no es suficiente con tener las capacidades cognitivas, sino que también son necesarias unas condiciones del entorno. Los llamados «derechos de segunda generación» (salud, educación, trabajo, vivienda...) pusieron de relieve que, si bien es importante proclamar la libertad individual de las personas, también lo es tener las condiciones para ejercerla. En esta dirección y diferenciándose de la concepción individualista de la autonomía personal, las versiones sociales y relacionales consideran que la plena autonomía sólo se alcanza en condiciones sociales de apoyo. La teoría del reconocimiento de Axel Honneth, por ejemplo, ha destacado la importancia del autorrespeto, la autoconfianza y la autoestima para ejercer la autonomía y la necesidad de estar atentos a las situaciones de relación con los demás en las cuales la autonomía es vulnerable. En la valoración de la capacidad de decisión de un paciente y en el proceso de información y de consentimiento para participar en un estudio clínico, por lo tanto, es imprescindible no sólo el reconocimiento jurídico del interlocutor, sino también el reconocimiento y acompañamiento afectivos.73 16 La necesidad de valorar con precisión la capacidad del paciente para dar el consentimiento aumenta a medida que lo hacen los riesgos y las molestias de la investigación clínica, especialmente cuando el estudio no aporta ningún beneficio directo para el paciente. Cuanto más elevados son los riesgos y las molestias, más elevado debe ser el grado de protección del paciente y de control de la investigación 16.1 No todos los estudios clínicos requieren extremar los procesos de protección con la misma intensidad. En la recomendación 12 se han señalado las razones que justifican que las exigencias éticas y procedimentales de estudios dirigidos a personas incapaces de dar el consentimiento informado sean más rigurosas. En este sentido, cuanta menos capacidad tiene una persona para otorgar su con- 73 ANDERSON, J.; HONNETH, A. (2004): «Autonomy, vulnerability and justice», en CHRISTMAN, J.;ANDERSON, J. (ed.): Autonomy and the challenges to liberalism, Cambridge, Cambridge University Press, p. 127-149. HONNETH, A. (1992): Kampf um Anerkennung (trad. castellana de M. Ballestero: La lucha por el reconocimiento. Por una gramática moral de los conflictos sociales, Barcelona, Crítica, 1997). 60 sentimiento informado, más importante es que el proceso de investigación no suponga riesgos y/o molestias y que le pueda aportar algún beneficio. Por lo tanto, un estudio clínico que comporte muchos beneficios y pocos riesgos y molestias requiere un estándar de competencia menor que la situación contraria, y si estos beneficios son directos (BD), un estándar de competencia menor que si los beneficios no son directos (BND). No todos los estudios clínicos requieren extremar los procesos de protección con la misma intensidad. Por ejemplo, y en general, los estudios clínicos de los que se esperan muchos beneficios para el paciente y los riesgos y las molestias son insignificantes requieren menos atención que un estudio clínico en el cual no se esperan beneficios directos, aunque los riesgos y molestias también sean leves, porque en una investigación de la cual no se espera ningún beneficio directo para el paciente es importante garantizar que los intereses científicos o incluso económicos no se impongan al bienestar del paciente. 16.2 La protección de los adultos incapacitados respecto a los menores de edad. En una primera aproximación y si nos atenemos al artículo 7.3.a del Real decreto 223/2004, de 6 de febrero, por el cual se regulan los ensayos clínicos con medicamentos, parece que en investigación clínica hay una menor protección jurídica de las personas adultas incapacitadas legalmente respecto de los menores de edad. Efectivamente, el citado artículo dice que en los casos en que el sujeto del ensayo sea un menor de edad «el promotor pondrá en conocimiento del ministerio fiscal las autorizaciones de los ensayos clínicos cuya población incluya a menores», y en cambio esta medida de protección no se prevé en el artículo 7.3.b del mismo Real decreto, dedicado a aquellas situaciones en que el sujeto es un adulto sin capacidad para otorgar el consentimiento informado. Esta diferencia fue parcialmente corregida en la Ley española 14/2007 de investigación bioética, que en los artículos 20.2.c (dedicado a las personas que no tienen capacidad para expresar su consentimiento y que participan en una investigación de la cual no se esperan beneficios directos para ellas) y 21.1.c (dedicado a la investigación en personas incapaces de consentir a causa de una situa- 61 ción clínica de emergencia) dice que una de las condiciones es «que la autorización de la investigación se ponga en conocimiento del ministerio fiscal» (con respecto a investigaciones clínicas de estas características, véanse las recomendaciones 12 y 13). Si consideramos que las personas sin capacidad para otorgar su consentimiento informado requieren el mismo grado de protección que la infancia y la adolescencia, este tratamiento diferenciado se podría justificar no en términos de mayor o menor protección, sino por el hecho de que la tutoría de una persona incapacitada ha sido otorgada en un proceso judicial que ha evaluado la competencia del tutor, un proceso que no sigue la paternidad natural. Sin entrar a considerar que en el ámbito de la infancia eso también se produce sin que sea motivo de excepción, hay que advertir que los tutores pueden sufrir, como todas las personas, procesos de transformación, y que se podría dar el caso de tutores que no velaran por los intereses de la persona incapaz. 17 Ética y jurídicamente, la capacidad de decisión de una persona siempre se presupone, la incapacidad debe demostrarse y en caso de duda prevalece la presunción de capacidad 17.1 La carga de la prueba sobre la incapacidad para autorizar o rechazar la participación en un estudio clínico recae en el médico responsable del reclutamiento, que por prudencia debería pedir la opinión de otro profesional, y en caso de desacuerdo con el paciente o con las personas que lo cuidan, será necesaria la intervención del juez. Una persona sólo puede ser considerada incapaz de tomar una decisión que le afecta después de que se haya hecho todo lo posible para ayudarla a tomarla y de haber evaluado su capacidad con instrumentos válidos y reconocidos (véase la recomendación 15). V. INFORMACIÓN 18 El protocolo del estudio clínico debe recoger y garantizar que se dará una información oral sin prisas, veraz, precisa, exhaustiva, comprensible, continuada y adaptada a las personas a las cuales va dirigida y con un intervalo de tiempo razonable para tomar la decisión. Hay que tener especial cuidado con la capacidad de comprensión de los pacientes y 62 también con la del tutor, curador o guardador de hecho, sobre todo si son de otros contextos culturales, y asegurarse de que han entendido las características del estudio y las consecuencias de participar en él. Si la persona que tiene que dar el consentimiento tiene dificultades para entender la lengua con la cual el investigador le informa, es necesario que haya un traductor imparcial. Con personas de contextos culturales muy diferentes del occidental, haría falta la presencia de un mediador cultural 18.1 La normativa vigente. Según las declaraciones internacionales y la legislación vigente, y como norma general, no se puede hacer ninguna intervención en una persona en materia de salud sin su consentimiento informado y libre. La información debe darse en una entrevista, en condiciones y formatos accesibles y apropiados a las capacidades y necesidades de la persona afectada, y después por escrito. La información debe ser veraz, precisa, exhaustiva y adecuada a las necesidades y requerimientos del paciente, tutor, curador y guardador de hecho, y deben tener el tiempo necesario para pensárselo y consultarlo con quien consideren conveniente.74 El intervalo de tiempo entre la información y la decisión facilita la valoración ponderada por parte del paciente o su representante legal. La información debe ser continuada y debe adquirirse el compromiso de informar sobre los hechos relevantes que se produzcan durante el transcurso del estudio y que puedan influir en la decisión de continuar participando en la investigación. 18.2 La importancia de la información oral. Sobre la importancia de la información oral, el Tribunal Supremo ha dicho lo siguiente: «La doctrina de esta Sala ha declarado con reiteración que la exigencia de la constancia escrita de la información tiene, para casos como el que se enjuicia, mero valor ad probationem (SSTS 2 octubre 1997; 26 enero y 10 noviembre 1998; 2 noviembre 2000; 2 julio 2002; 74 Art. 7 del Pacto internacional de los derechos civiles y políticos (1966/1976). Norma 4.8.7 de la Guía de buena práctica clínica CPMP/ICH/95. Arts. 4, 5 y 8 de la Ley española 41/2002. Comentario sobre la pauta 4 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). Art. 7 del RD 223/2004. Art. 13.1 del Protocolo adicional al Convenio para la protección de los derechos humanos (2005). Art. 6 de la Declaración universal sobre bioética y derechos humanos (2005). Arts. 4, 15 y 20.1 de la Ley española 14/2007. Art. 24 de la Declaración de Helsinki (2008). 63 29 julio 2008), garantizar la constancia del consentimiento y de las condiciones en que se ha prestado, pero no puede sustituir a la información verbal, que es la más relevante para el paciente, [...]». También ha señalado reiteradamente que «la información al paciente ha de ser puntual, correcta, veraz, leal, continuada, precisa y exhaustiva, es decir, que para la comprensión del destinatario se integre con los conocimientos a su alcance para poder entenderla debidamente y también ha de tratarse de información suficiente que permita contar con datos claros y precisos para poder decidir [...]».75 18.3 Reclutamiento de personas de contextos culturales diferentes o de personas con poca formación y pocos recursos sociales y comunicativos. Debe prestarse mucha atención a los procesos de reclutamiento de personas de contextos culturales diferentes o de personas con poca formación y pocos recursos sociales y comunicativos. «El investigador debe tener en mente que la capacidad del potencial sujeto para comprender la información necesaria para dar su consentimiento depende de la madurez, inteligencia, educación y sistema de creencias del individuo. Depende, además, de la capacidad del investigador y de su buena voluntad para comunicar con paciencia y sensibilidad.»76 No debería reclutarse a ninguna persona con la cual no sea posible comunicarse fluidamente, o con su tutor, curador o guardador de hecho. La comunicación fluida hace referencia a la lengua y a los parámetros culturales. 18.4 La calidad de la información. Algunos déficits de comprensión están relacionados con la baja calidad del procedimiento y de la información que se da. Hay mucha literatura que describe el éxito de métodos para aumentar la capacidad de comprensión y decisión; por ejemplo, estructurar bien la información, preparar las entrevistas informativas, hacer buenos resúmenes, disponer de tiempo para la entrevista, reforzar la explicación con apoyo grá- 75 Tribunal Supremo. Sala Civil. Resolución n.º 2/2009, de 21 de enero (sentencia sobre responsabilidad civil médica) y Resolución n.º 1216/2007, de 28 de noviembre (sentencia sobre responsabilidad médica), respectivamente. 76 Comentario sobre la pauta 4 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). 64 fico (vídeos, diapositivas, fotografías, dibujos), retroalimentación, etc.77 El investigador que informa de las características del estudio clínico e invita a los afectados a participar debería comprobar, con respeto y tacto, que sus interlocutores han entendido aquello que les ha comunicado y que están en condiciones de tomar una decisión. 19 Es necesario informar oralmente y por escrito, y siempre que sea procedente, de las treinta y siete cuestiones que se señalan en esta recomendación, algunas de las cuales se amplían en recomendaciones aparte 19.1 Según la deontología, las declaraciones internacionales y la legislación vigente, antes de obtener el consentimiento, debería proporcionarse al paciente, tutor, curador o guardador de hecho la información siguiente:78 1) Que se le invita a participar en el estudio y las razones para hacerlo. 2) Que la participación es absolutamente voluntaria y que puede negarse a participar y retirarse del estudio en cualquier momento sin tener que dar explicaciones y sin que eso repercuta de ninguna manera en la calidad de su asistencia sanitaria (véase la recomendación 24). 77 SIMÓN LORDA, P. (2004): «El consentimiento informado: alianza y contrato, deliberación y decisión», en COUCEIRO, A. (2004): Ética en cuidados paliativos, Madrid, Triacastela, p. 79-108. Una recopilación bibliográfica se puede encontrar en el capítulo "Métodos para aumentar la capacidad" del artículo de DILIP, V.; JESTE, M.D.; SAKS, E.S. (2006): «Decisional capacity in mental illness and substance use disorders: Empirical database and policy implications», o.c. Sobre la eficacia de la mejora de los procedimientos de autorización (enhanced consent procedures) y del consentimiento con experiencia (experienced consent), MITTAL, D.; PALMER, B.W.; DUNN, L.B.; LANDAS, R.; GHORMLEY, C.; BECK, C.; GOLSHAN, S.; BLEVINS, D.; JESTE, D.V. (2007): «Comparison of two enhanced consent procedures for patients with mild Alzheimer disease or mild cognitive impairment», American Journal of Geriatric Psychiatry (febrero 2007), 15 (2), p. 163-167, y OLDE RIKKERT, M.G.M.; VAN DEN BERCKEN, J.H.L. (1997): «Experienced consent in geriatrics research: a new method to optimize the capacity to consent in frail elderly subjects», Journal of Medical Ethics, 1997, 23, p. 271-276. 78 Pauta 3.3 de las Pautas para la buena práctica clínica (BPC) en ensayos con productos farmacéuticos (OMS-1995). Norma 4.8.10 de la Guía de buena práctica clínica CPMP/ICH/95. Art. 4 de la Ley española 41/2002. Art. 3.2 de la Directiva 2001/20/CE. Comentario sobre la pauta 4 y la pauta 5 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). Art. 7 del RD 223/2004. Art. 13.2 del Protocolo adicional en el Convenio para la protección de los derechos humanos (2005). Art. 21 de la Declaración universal de bioética y derechos humanos (2005). Art. 15 de la Ley española 14/2007. Arts. 24 y 33 de la Declaración de Helsinki (2008). 65 3) 4) 5) 6) 7) 8) 9) 10) 11) 12) 13) 14) 15) 16) 17) 18) 19) 20) Que puede hacer todo tipo de preguntas relacionadas con la investigación al responsable o colaboradores del estudio. Que si quiere, antes de dar el consentimiento se tome el tiempo necesario para consultarlo con otras personas (familiares, amigos, médico de cabecera...). Los objetivos del estudio. Identificar y explicar el procedimiento experimental (fármaco, intervención quirúrgica, grupo terapéutico, prueba pronóstica, entrevista clínica...). La denominación genérica del producto de estudio. El número de participantes previstos. Las condiciones de inclusión, de exclusión y de finalización anticipada de la investigación. Los centros y/o países que está previsto que participen en él. El tipo y las características del diseño del estudio (p. ej., aleatoriedad, doble ciego, placebo...). En estudios aleatorizados, la probabilidad de asignación en cada grupo de tratamiento. En estudios con placebo, en qué consiste. En estudios con placebo, la posible evolución de la patología durante el periodo en que el participante recibirá placebo. Los beneficios que se esperan obtener (directos y/o indirectos). Los riesgos y molestias para el paciente, diferenciando si hace falta entre los R-MBD y los R-MBND (véase la recomendación 10). Las medidas previstas para reaccionar a eventuales acontecimientos adversos y a las preocupaciones de los participantes en la investigación. Los procedimientos preventivos, diagnósticos y terapéuticos alternativos (habituales) disponibles y las ventajas e inconvenientes con respecto a lo que se propone en el estudio clínico. Cuál es la duración esperada de la participación del paciente y, si procede, del acompañante. Los procedimientos que requiere el estudio (número de visitas, tipo y número de exploraciones...) y las diferencias con la atención médica habitual (si hay más visitas, más pruebas y/o si son diferentes...). 66 21) Las circunstancias y/o razones previsibles que pueden motivar la finalización de la participación en el estudio. 22) Que de acuerdo con la legislación vigente, después de la realización del ensayo clínico no se le podrá seguir administrando la medicación en estudio si ésta no ha sido autorizada por la administración sanitaria como especialidad farmacéutica con esta indicación o condiciones de uso. Y que podría ocurrir que aunque esta medicación sea autorizada, el Sistema Nacional de Salud decida no financiar su uso. 23) Cuáles son las responsabilidades del participante y el tutor, curador o guardador de hecho. 24) Las disposiciones tomadas para garantizar el respeto a la intimidad de las personas y la confidencialidad de los datos de carácter personal, de acuerdo con las exigencias previstas en la legislación, tanto en el propio país como en otros en caso de transferencia de datos. 25) Del proceso de cancelación o destrucción de los datos de carácter personal cuando hayan dejado de ser necesarias o pertinentes para la finalidad para la cual fueron solicitados o registrados y en los plazos que establece la ley. 26) Las personas que tendrán acceso directo a los datos personales (monitores, auditores, CEIC y autoridades competentes). 27) Que tiene derecho de acceso, rectificación, cancelación y oposición de sus registros de la base de datos del estudio, y la persona o lugar adonde se tiene que dirigir para ejercerlo. 28) Que se les informará de los hechos relevantes que surjan durante el transcurso del estudio que puedan influir en la decisión de continuar participando en la investigación y, si quieren, de los resultados finales siempre que sea posible y pertinente para la salud del participante. 29) Del compromiso que, cuando se publiquen los resultados, si el interesado quiere se le facilitará una copia o el acceso a estos resultados.79 79 Art. 27.2 de la Ley española 14/2007 de investigación biomédica. Hay que mencionar aquí el artículo 5.5 de esta Ley, que dice: «Si no fuera posible publicar los resultados de una investigación sin identificar a la persona que participó en la misma o que aportó muestras biológicas, tales resultados sólo podrán ser publicados cuando haya mediado el consentimiento previo y expreso de aquélla». 67 30) Quiénes son los patrocinadores de la investigación, el origen de la financiación, la propiedad y utilización posterior (sobre todo comercial) de los resultados de la investigación y de los datos o de los materiales biológicos, y de la afiliación institucional de los investigadores. En los estudios sin beneficios directos para el paciente se suele apelar a la solidaridad, al progreso de la ciencia y al bienestar de la humanidad. En estos casos más que nunca, el paciente, tutor, curador o guardador de hecho deben ser informados de si el estudio tiene, o no, intereses comerciales y de quién es la propiedad. No se puede permitir apelar a las motivaciones altruistas de los participantes sin explicar también las motivaciones de los promotores. 31) Si el participante recibirá o no una compensación económica y las condiciones de ésta (el reintegro de los gastos extraordinarios o pérdidas de productividad que se prevean por la participación en el estudio). (Véase la recomendación 30.) 32) Si el centro sanitario y/o los profesionales recibirán, o no, alguna compensación económica por la tarea realizada en el estudio clínico. 33) En caso de ensayo clínico, referencia a la contratación y cobertura de una póliza de seguro de responsabilidad civil por los posibles perjuicios que pueda causar el ensayo (con el nombre de la compañía y el número de póliza). 34) Los posibles conflictos de intereses. 35) Si se obtienen muestras biológicas: a) de la anonimización, si procede; b) que después de la anonimizacióm ya no será posible ejercer el acceso, rectificación, cancelación y oposición; c) que la muestra puede ser cedida o utilizada por terceros (p. ej., laboratorios externos) o no, y el proceso de autorización; d) si se tiene previsto destruirlas cuando se acabe la investigación y, si no es así, los detalles sobre su almacenaje (dónde, cómo, por cuánto tiempo y su disposición final); e) posible uso futuro, y que tienen el derecho de decidir sobre este uso futuro, de hacer destruir el material y de negarse a su almacenaje. 68 36) Que un Comité Ético de Investigación Clínica ha aprobado o autorizado el protocolo de investigación (con los datos de contacto) o la comisión de investigación o similar, si procede. 37) El nombre del investigador principal del centro y los datos de contacto del responsable. 20 La hoja de información para el paciente debe estar redactada de manera veraz, precisa, exhaustiva, comprensible y adaptada a las capacidades de las personas a las que va dirigida 20.1 El consentimiento informado y el documento de consentimiento informado. El consentimiento informado es el acto de información por parte del profesional, de conversación entre las partes y de autorización a participar en el estudio clínico por parte del paciente o tutor. Esta autorización puede quedar plasmada o no en un documento de consentimiento informado, el escrito en el cual se recoge la información que da el profesional y la autorización que otorga el paciente o tutor. El documento de consentimiento informado tiene dos partes diferenciadas: la hoja de información y la hoja de consentimiento por escrito. La hoja de información suele tener más de una página, recoge todo aquello significativo del estudio de una manera clara y entendedora (véase la recomendación 20) y puede haber una para el paciente y otra para el tutor (véase la recomendación 21). En la hoja de consentimiento por escrito, que suele tener sólo una página, el paciente —o si está incapacitado legalmente, su representante legal— declara y firma que ha sido informado correctamente y que autoriza la participación en el estudio clínico (véanse las recomendaciones del capítulo VI. CONSENTIMIENTO). El paciente o su representante legal tienen que tener una copia de las dos hojas.80 Según la legislación vigente, la hoja de información al paciente o a su tutor debe contener toda la información necesaria y debe estar redactada de manera veraz, precisa, exhaustiva, comprensible y adaptada a las capacidades de aquellos a quienes va dirigida.81 80 Norma 4.8.11 de la Guía de buena práctica clínica CPMP/ICH/95. 81 Arts. 4, 5 y 8 de la Ley española 41/2002. Art. 5 de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo. 69 20.2 Sobre la veracidad de la información y la posibilidad de cierto grado de engaño en aquellas investigaciones en que esto es necesario. Tal como señala la pauta 6 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002), mentir a los sujetos es una táctica poco utilizada en la investigación biomédica y más usual en investigadores sociales y conductuales, que algunas veces informan expresamente de manera engañosa a los sujetos para estudiar sus actitudes y comportamiento. Las condiciones que deberían reunir este tipo de estudios, que son excepcionales, son las siguientes: i) ii) iii) iv) v) vi) vii) viii) ix) que no es posible ningún otro método de investigación; que no hay ningún tipo de riesgo ni molestia para los participantes; que se analiza cuidadosamente el hecho de que los participantes pueden ofenderse por no haber sido informados, al enterarse de que han participado en un estudio con condiciones falsas; que se obtendrán avances significativos gracias a la investigación; que el estudio clínico reúne las condiciones científicas; que nada de lo que se omite en la información ocasionaría, si se supiera, que una persona razonable rehusara participar en él; una vez acabado el proceso con cada una de las personas, informar adecuadamente sobre el engaño a los participantes; considerar la desaprobación de las personas que han sido engañadas como efectos adversos que pueden aconsejar suspender el estudio; que la persona puede no autorizar que el investigador use la información obtenida. 21 Además de la hoja de información dirigida a los tutores, a los pacientes con dificultades de comprensión, apreciación y razonamiento debe facilitárseles una hoja de información resumida de fácil comprensión o cualquier otro apoyo informativo adaptado a sus capacidades, a fin de que puedan intervenir tanto como sea posible en la toma de decisiones 21.1 Si, tal como señala una buena práctica clínica y la legislación vigente, las personas incapaces de otorgar el consentimiento deben participar en la medida de lo posible en el proceso de información y decisión, las investigaciones dirigidas a este sector de población 70 deben disponer, además de la hoja de información convencional, de una hoja de información o de cualquier otro apoyo (p. ej., un vídeo) adaptado a sus características y capacidades. En este sentido, el artículo 4.1 de la Ley española 14/2007 de investigación biomédica dice: «La información se prestará a las personas con discapacidad en condiciones y formatos accesibles apropiados a sus necesidades.» Corresponde al promotor del estudio facilitar los procedimientos y los instrumentos que permitan a los profesionales y tutores hacerlo posible. 22 En estudios clínicos con riesgos y molestias derivados de un tratamiento experimental con posibles beneficios directos (R-MBD), pero también con riesgos y molestias de pruebas complementarias sin beneficios directos para el paciente (R-MBND), debería disponerse de una hoja de información y de un consentimiento por escrito para el tratamiento experimental y otro para las pruebas complementarias 22.1 En la recomendación 11 se ha señalado que actualmente no es posible que los CEIC emitan dos dictámenes sobre un mismo protocolo de investigación, por ejemplo autorizando el tratamiento experimental y rechazando los estudios complementarios, y que sería deseable cambiar esta situación. Si eso fuera posible y con respecto al consentimiento informado, en estudios clínicos con riesgos y molestias derivados de un tratamiento con posibles beneficios directos para el paciente (R-MBD), pero también con riesgos y molestias de pruebas complementarias sin posibles beneficios directos (R-MBND), debería haber una hoja de información y una hoja de consentimiento por escrito para el tratamiento experimental, y una hoja de información y una hoja de consentimiento por escrito para las pruebas complementarias, con el fin de facilitar la posibilidad de autorizar la participación en el tratamiento pero no en las pruebas complementarias. 23 Si se pretende hacer un análisis genético, es necesario proporcionar información expresa y específica y el consentimiento correspondiente 23.1 Tal como señala la legislación vigente, es necesario el consentimiento expreso y específico por escrito —y evidentemente oral— para la realización de un análisis genético.82 82 Art. 48.1 de la Ley española 14/2007. 71 24 Quien firma el consentimiento informado no debe tener ningún tipo de duda sobre el hecho de que puede negarse a participar en la investigación y retirarse en cualquier momento sin necesidad de dar explicaciones y sin que eso repercuta de ninguna manera en su asistencia y relación sanitaria 24.1 El médico y la hoja de información deben informar esmeradamente al paciente, tutor, curador o guardador de hecho que la negativa a participar en la investigación o la decisión de retirarse por cualquier motivo nunca puede suponer ningún tipo de desventaja, ni de perjuicio, ni perturbar su atención, y que pueden hacerlo en cualquier momento y sin tener que dar explicaciones.83 25 La información oral y escrita debería advertir que hay que prestar atención a las posibles relaciones de dependencia del paciente, tutor, curador o guardador de hecho con el profesional sanitario que les propone participar en la investigación, a fin de que esta situación no influya en la decisión 25.1 El artículo 26 de la Declaración de Helsinki (2008) insiste en el hecho de que «Al pedir el consentimiento informado para la participación en la investigación, el médico debe poner especial cuidado cuando el individuo potencial está vinculado con él por una relación de dependencia o si consiente bajo presión. En una situación así, el consentimiento informado debe ser pedido por una persona calificada adecuadamente y que nada tenga que ver con aquella relación». La cuestión de la posible relación de dependencia es especialmente delicada, porque la exigible buena relación terapéutica entre el profesional sanitario y el paciente, tutor, curador o guardador de hecho genera, como es natural, una relación de confianza y agradecimiento. El consentimiento libre significa que no hay coacción de ningún tipo, ni siquiera un consentimiento motivado por el sentimiento de deuda o gratitud. Cuando esto influye en la decisión, podemos hablar de «poblaciones cautivas». 83 Art. 7.5 del RD 223/2004, de 6 de febrero, por el cual se regulan los ensayos clínicos con medicamentos. Art. 6 de la Declaración universal sobre bioética y derechos humanos (2005). Arts. 4.3, 4.4 y 14.4 de la Ley española 14/2007. Arts. 24 y 34 de la Declaración de Helsinki (2008). 72 Es evidente que la mejor manera de evitar con garantías las relaciones de dependencia en el reclutamiento en estudios clínicos es que el profesional sanitario no participe en las investigaciones en las cuales sus pacientes pueden ser reclutados o han sido reclutados. Y no sólo para alejar del todo la posibilidad de «pacientes cautivos», sino también de «profesionales sanitarios cautivos» con la investigación. En este sentido, no es desmesurado considerar —y lamentablemente la historia de la investigación clínica lo atestigua—84 que puede haber situaciones en las cuales el interés personal del profesional en el estudio clínico provoque un sesgo en su protección del paciente y en su independencia en el momento de presentar la investigación y aconsejar la participación o no.85 Ahora bien, eso requeriría un esfuerzo económico y organizativo desmesurado que reduciría significativamente la investigación clínica. Mientras tanto, consideramos una buena medida que las hojas de información y de consentimiento por escrito adviertan de este peligro y que las asociaciones de afectados se conviertan también en una fuente de contraste y de información (véanse las recomendaciones 4 y 29). 26 Cuando no se espera ningún beneficio directo para el paciente, en el proceso de información oral hay que garantizar que las personas afectadas no tienen una «falsa idea terapéutica» o «equívoco terapéutico» (therapeutic misconception) 84 La literatura sobre los horrores en la historia de los estudios clínicos es muy amplia. A modo de ejemplo: BRODY, H. (1998): The ethics of biomedical research, Oxford, Oxford University Press. ROTHMAN, D. (1991): Strangers at the bedside, New York, Basic Books. ANNA, G.; GRODIN, M. (1992): The Nazi Doctors and the Nuremberg Code, Oxford, Oxford University Press. LUNA, F. (2001): Ensayos de bioética. Reflexiones desde el Sur, México, Fontamara. Hay que destacar que las aberraciones de la época nazi sólo son una parte de una larga historia de abusos, que llegan hasta la segunda mitad del siglo XX. Por ejemplo, en la obra de WEYERS, W. (2003): The abuse of man. An illustrated history of dubious medical experimentation (Ardor Scribendi, LTD, New York), la medicina nazi ocupa 2 de los 26 capítulos del libro (73 páginas de las más de 750 que tiene la obra). 85 Sobre el interés desmesurado de algunos profesionales sanitarios por publicar, conseguir brillantes descubrimientos, aumentar el prestigio o recibir incentivos de la industria farmacéutica, véase: EMANUEL, E.; STEINER, D. (1995): «Institutional conflict of interest», New England Journal of Medicine, vol. 332, 1995, p. 262-267. WEATHERALL, D. (2000): «Academia and industry: increasingly uneasy bedfellows», The Lancet, vol. 355, 2000, p. 1574. BODENHEIMER, TH. (2000): «Uneasy alliance. Clinical investigators and the pharmaceutical industry», New England Journal of Medicine, vol. 342, 2000, p. 1539-1544. 73 26.1 El profesional que da la información debe comprobar que el paciente, tutor, curador o guardador de hecho no tiene una «falsa idea terapéutica» o «equívoco terapéutico» (therapeutic misconception), es decir, que el paciente o las personas que otorgan el consentimiento por representación consideran de alguna manera que recibirá un tratamiento que le ayudará, cuando en realidad el posible beneficio que se obtendrá será únicamente para la investigación médica.86 27 Cuando el beneficio directo previsto sea igual o menor que los riesgos y molestias y el consentimiento es por representación, la información oral y escrita debería advertir que hay que prestar atención en la influencia que puede tener la posible desazón de impotencia ante la enfermedad o incluso de deuda con el paciente, y recordar la necesidad de una decisión adecuada a las circunstancias y proporcionada a las necesidades del paciente 27.1 No sólo no debe generarse una falsa idea terapéutica (véase la recomendación 26), sino que también debe procurarse que el tutor, curador o guardador de hecho no se aferren incondicionalmente a cualquier intervención sanitaria fruto de la no aceptación de la enfermedad o de la situación, o de una desazón de impotencia ante la enfermedad, o de un sentimiento de deber, o incluso de culpa o deuda no satisfecha con el paciente, por ejemplo por no haber actuado en su momento, o para compensar otros déficits, o para tranquilizar remordimientos... En estas situaciones, sería conveniente que el profesional sanitario y la hoja de información advirtieran del peligro de dejarse llevar por estos sentimientos. 28 Los pacientes, tutores, curadores o guardadores de hecho deberían disponer, además, de una guía que informe de lo que significa participar en un estudio clínico y que les oriente y facilite el proceso de decisión 86 APPELBAUM, P. LOREN, H.R.; LIDZ, CH.W.; BENSON, P.; WINSLADE, W. (1987): «False hopes and best data: consent to research and the therapeutic misconception», The Hasting Center Report, vol. 12, n.º 2, p. 20. JOFFE, S. [et al.] (2001): «Quality of informed consent in cancer clinical trials: a cross-sectional survey», The Lancet, vol. 358, p. 1772-1777. BERTOLI, A.M. (2007): «Lack of correlation between satisfaction and knowledge in clinical trials participants: A pilot study», Contemporary Clinical Trials, 23 de mayo de 2007. 74 28.1 La mayoría de las veces no es fácil decidir participar o no en una investigación clínica. Y en algunas ocasiones se convierte en una cuestión extraordinariamente difícil; por ejemplo, cuando debe tomarse una decisión que afecta a otra persona y los beneficios esperados son inciertos y los riesgos y las molestias elevados. Por otra parte, la hoja de información da por hecho que las personas saben o tienen facilidad para entender qué es un estudio clínico, para qué sirve y qué significa participar en él, lo cual no siempre es así. En estos casos no es suficiente con una buena información oral y escrita sobre las características del estudio y sobre los resultados, riesgos y molestias esperados. La información científica es importantísima, pero aquello que situará a las personas afectadas en una situación problemática no es, o no tendría que ser, la comprensión del estudio, sino la valoración de si vale la pena participar en él o no. Por lo tanto, habría que ofrecer una ayuda neutral para abordar este proceso en aquellos estudios que presentan un cierto grado de dificultad ética. La razón por la cual los protocolos de investigación clínica incluyen una buena información sobre las características del estudio y nunca una guía que oriente el proceso de decisión parece obvia: el facultativo, y menos aún los promotores del estudio, no pueden influir de ninguna manera en la decisión de los afectados y, por lo tanto, deben limitarse a dar una buena información sobre el estudio. Ahora bien, hay tres razones que aconsejan una ayuda neutral para abordar el proceso de decisión en aquellos estudios que presenten un cierto grado de dificultad ética: i) En algunos estudios clínicos intervienen variables imprecisas, o equiparables pero contradictorias, lo cual hace que las personas que deben tomar la decisión se encuentren en una situación éticamente compleja. ii) En el ámbito sanitario, el consentimiento informado es un acto de reconocimiento de la dignidad, la libertad y la responsabilidad del paciente.Ahora bien, y tal como se ha señalado en muchas ocasiones, no puede suponer en ningún momento el abandono del paciente o personas próximas ante situaciones que son nuevas para ellos, que sufren de manera directa y que a veces no disponen de todas las habilidades necesarias para abordarlas. 75 iii) Muy a menudo el paciente, tutor, curador o guardador de hecho pide consejo al facultativo, que suele tener un buen conocimiento científico del estudio, pero no siempre de las variables éticas que intervienen en él. Esta guía también contribuiría a promover la importancia de la investigación en el bienestar de la sociedad y a ampliar la participación de la comunidad en este asunto.87 29 Las asociaciones de afectados deberían estar informadas de los estudios clínicos que se realizan y ofrecer un servicio de apoyo a las personas que se plantean participar en ellos 29.1 Las asociaciones de afectados deberían estar informadas de los estudios clínicos que se realizan en su ámbito de influencia. Eso posibilitaría que los pacientes, tutores, curadores o guardadores de hecho dispusieran, si quieren, de otro punto de información y apoyo en el proceso de decisión. En este sentido, hay que recordar que la Directiva 2001/20/CE del Parlamento Europeo y del Consejo establece, después de decir que el participante en el ensayo o su representante legal deben recibir la información necesaria del investigador, que «los participantes deben disponer de un punto de contacto, donde puedan obtener más información» (art. 3.4). 30 No se puede dar ningún incentivo o estímulo económico; sólo se pueden retribuir los gastos generados por participar en la investigación. Si en el transcurso del estudio se da algún regalo, en ningún caso puede tener un valor persuasivo y este gesto debería ser aprobado por el CEIC 30.1 La normativa. La legislación vigente prohíbe cualquier forma de estímulo económico para participar en un estudio clínico. Sólo permite el reintegro de los gastos extraordinarios y pérdidas de productividad que deriven de la participación en el ensayo.88 Tal como señala la Ley española 14/2007, «Las personas que hayan sufrido daños como consecuencia de su participación en un pro- 87 Posición 10 de la Position Statement Informed Consent for Research on Human Subjects with Dementia American Geriatrics Society Ethics Committee (2007). 88 Art. 5.d de la Directiva 2001/20/CE. Art. 3.8 del RD 223/2004. Art. 15 de la Declaración universal sobre bioética y derechos humanos (2005). 76 yecto de investigación recibirán la compensación que corresponda» (art. 18.1). «Se presume, salvo prueba en contra, que los daños que afecten a la salud de la persona sujeta a la investigación, durante su realización y en el año siguiente a su terminación, se han producido como consecuencia de la investigación. Sin embargo, una vez concluido el año, el sujeto de aquélla estará obligado a probar el daño y el nexo entre la investigación y el daño producido» (art. 18.4). 30.2 Regalos. En algunas investigaciones, los promotores hacen algún presente a las personas que participan en ellas. Estos actos tendrían que ser previamente autorizados por el CEIC y en ningún caso pueden suponer un incentivo. VI. CONSENTIMIENTO 31 Cuando el paciente está incapacitado legalmente, debe obtenerse autorización de su representante legal. El paciente debe intervenir tanto como sea posible en el proceso informativo y en la toma de decisiones. Debe respetarse su rechazo o asentimiento y si su capacidad no permite expresarlo, debe tenerse en cuenta su presunta voluntad 31.1 Según las declaraciones internacionales y la legislación vigente, cuando el paciente no es competente para autorizar su participación en un estudio clínico:89 i) Debe obtenerse la autorización de su representante legal. ii) El paciente no debe oponerse a la participación en el estudio o previamente no debe haberse manifestado en contra.90 89 Art. 7.3 del RD 223/2004. Arts. 4 y 13 de la Ley española 14/2007. Art. 5 de la Directiva 2001/20/CE. Pauta 15 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). Art. 7 de la Declaración universal sobre bioética y derechos humanos (2005). 90 Art. 17.1v del Convenio de Oviedo (1997). Art. 11 de la Ley española 41/2002. La posición 9 de la Position Statement Informed Consent for Research on Human Subjects with Dementia American Geriatrics Society Ethics Committee (2007) dice: «En general, debe respetarse la negativa de un sujeto (potencial), aunque haya perdido la capacidad de toma de decisiones, La única excepción son aquellos protocolos que cumplan las cuatro condiciones siguientes: 1) que haya un alto potencial de beneficio en relación con el riesgo; 2) que el acceso a este beneficio sólo se pueda obtener a través de este estudio o investigación (p. ej., un prometedor fármaco experimental en agitación grave, en sujetos con demencia que no han obtenido resultados con los tratamientos convencionales); 3) que se obtenga consentimiento expreso por poderes, y 4) que se tengan en cuenta las precauciones adicionales señaladas por el comité de ética.» 77 iii) El paciente debe intervenir tanto como sea posible en la toma de decisiones. «Cuando las condiciones del sujeto lo permitan, éste deberá prestar además su consentimiento para participar en el ensayo [...]. En este caso, el investigador deberá tener en cuenta la voluntad de la persona incapaz de retirarse del ensayo.»91 iv) El consentimiento debe reflejar la presunta voluntad del participante. 31.2 La participación en la toma de decisiones del paciente incapaz culmina también en el hecho de que también firma —cuando tiene capacidad para hacerlo— la hoja de consentimiento por escrito.92 32 Cuando el paciente está incapacitado legalmente no siempre prevalece la decisión del tutor sobre la participación o no en el estudio clínico 32.1 La decisión del tutor no siempre puede ser la correcta. Cuando a través de un proceso judicial se ha evidenciado que una persona no es capaz de autogobernarse y por lo tanto se ha nombrado un tutor que ejerza la defensa de sus derechos, la responsabilidad de evaluar la conveniencia o no de participar en un estudio clínico recae en el tutor. Ahora bien, a pesar de esta responsabilidad que el juez ha depositado en el tutor, su decisión no siempre puede ser la correcta. Las tablas 2 y 3 (y la 4 y la 5, que están en la recomendación 36) pretenden facilitar y concretar el proceso de decisión en las diferentes situaciones que indican. Sobre ésta y las demás tablas hay que hacer las siguientes advertencias: i) se refieren a situaciones en las cuales no hay documento de instrucciones previas; ii) el «sí» y «no» señalan la voluntad del paciente (cuando la puede expresar) o del tutor respecto de participar en la investigación o no hacerlo, y la decisión que consideramos más correcta después de la valoración. Tal como hemos visto en la recomendación 31, la ley señala que el paciente no debe oponerse a ello, ya sea en el momento de la decisión o en un documento de instrucciones previas; 91 Art. 7.3.b.2 del RD 223/2004. 92 Norma 4.8.12 de la Guía de buena práctica clínica CPMP/ICH/95. 78 iii) las casillas en gris indican situaciones (A, B, C...) con alguna posible dificultad en la decisión, que es abordada posteriormente; iv) a pesar de la nítida delimitación de una tabla, en ética los márgenes de cada cuadro y la lógica binaria del «sí» y el «no» deben interpretarse a veces de una manera borrosa y difusa, y, sobre todo, atendiendo a las variables que intervienen en cada situación concreta. 32.2 La decisión en estudios clínicos con posibles riesgos y molestias (R-M), de los cuales se espera obtener un beneficio directo (BD) para pacientes incapacitados legalmente. En estos casos y siempre que se cumplan todos los requisitos señalados por la ley y en estas recomendaciones, que habrán sido evaluados y aprobados por un CEIC, se pueden dar las siguientes situaciones: TABLA 2 BD ≈ R-M BD > R-M93 BD < R-M94 Paciente incapacitado legalmente sí no sí no sí no sí no sí no sí no Tutor sí sí no no sí sí no no sí sí no no Decisión final que consideramos más prudente sí sí/ no sí no sí no no no sí no no no A B C D E F Situaciones susceptibles de profundización El «sí» y el «no» indican la voluntad o la decisión de la persona o personas respecto a participar o no en el estudio clínico. Situaciones B, C y E. El paciente quiere participar en un estudio con muchas posibilidades de que le sea beneficioso, y el tutor considera que no debería hacerlo (B). El paciente no quiere participar en un estudio en el cual no es posible valorar si prevalecen los 93 Tal como se ha señalado en la tabla 1: BD > R-M significa que los beneficios directos esperados (BD) son superiores a los riesgos y molestias previstos (R-M); BD ≈ R-M, que los beneficios directos esperados son equiparables a los riesgos y molestias previstos; y BD < R-M, que los beneficios directos esperados son inferiores a los riesgos y molestias previstos. 94 Con las condiciones descritas en la recomendación 12. 79 beneficios directos o los riesgos y/o molestias, y el tutor considera que sí debería hacerlo (C). El paciente no quiere participar en un estudio en el cual los riesgos y/o las molestias previstos son superiores a los beneficios directos esperados, y el tutor considera que sí debería hacerlo (E). En estas tres situaciones es difícil encontrar alguna razón aceptable que aconseje no atender la opinión del paciente. Situaciones A, D y F. El paciente no quiere participar en un estudio con muchas posibilidades de que le sea beneficioso, y el tutor considera que sí debería hacerlo (A). El paciente quiere participar en un estudio en el cual no es posible valorar si prevalecen los beneficios directos o los riesgos y/o molestias, y el tutor considera que no debería hacerlo (D). El paciente quiere participar en un estudio en el cual los riesgos y/o las molestias previstos son superiores a los beneficios directos esperados, y el tutor considera que no debería participar en él (F). Con respecto a la situación A, el artículo 17.1v del Convenio de Oviedo dice que sólo se puede hacer investigación en una persona que no tenga capacidad de dar el consentimiento si ésta no se opone a ello. Consideramos que a pesar de esta directriz y del carácter predictivo de todo estudio clínico, puede haber situaciones en las cuales no sea recomendable atender la opinión de la persona incapacitada, por ejemplo en aquellas en las que no hay tratamiento convencional, o el tratamiento convencional no consigue mejoras significativas en el paciente, y hay posibilidades fundamentadas de que el tratamiento experimental sí que lo consiga. Sobre las situaciones D y F, una razón que puede aconsejar no aplicar el criterio de atender la decisión de la persona afectada es que hacerlo puede generar un perjuicio al paciente y al tutor. 32.3 La decisión en estudios clínicos con posibles riesgos y molestias (R-M), de los cuales no se espera obtener ningún beneficio directo (BND) para pacientes incapacitados legalmente. En estos casos y siempre que se cumplan todos los requisitos señalados por la ley y en la recomendación 12, que habrán sido analizados y aprobados por un CEIC, se pueden dar las situaciones siguientes: 80 TABLA 3 BND ≈ R-M BND > R-M95 Paciente incapacitado legalmente sí no sí no sí no sí no Tutor sí sí no no sí sí no no Decisión final que consideramos más prudente sí no no no sí no no no G H I J Situaciones susceptibles de profundización BND < R-M Estos tipos de estudio tienen que ser rechazados por el CEI y, por lo tanto, no llegan a los pacientes ni tutores Situaciones G e I. En aquellas situaciones en las cuales el paciente no quiere participar en un estudio sin beneficios directos y el tutor considera que sí debería hacerlo, la ética y la ley señalan que debe prevalecer la voluntad del paciente, porque los beneficios para futuros pacientes y para la ciencia no pueden justificar los riesgos y posibles molestias para una persona que, a pesar de estar incapacitada, manifiesta su negativa a participar en un estudio. Situaciones H y J. En estas situaciones, en las que el paciente manifiesta su voluntad de participar en el estudio y el tutor considera que es mejor que no lo haga, y tratándose de estudios de los que no se espera ningún beneficio directo para el paciente y en cambio sí molestias y riesgos (que también repercutirán de alguna manera en aquellos que le cuidan) y que el paciente no es capaz de entender, apreciar, razonar y decidir correctamente, es juicioso considerar que debe prevalecer la decisión del tutor. 33 Cuando el paciente está incapacitado legalmente, tiene una patología que no ha hecho posible la manifestación de instrucciones previas y no se prevén beneficios directos del estudio clínico (BND) y sí riesgos y molestias, antes de incluirlo habría que indagar si la autorización se da 95 Tal como se ha señalado en la tabla 1: BND > R-M significa que los beneficios no directos esperados (BND) son superiores a los riesgos y molestias previstos (R-M); BND ≈ R-M, que los beneficios no directos esperados son equiparables a los riesgos y molestias previstos; y BND < R-M, que los beneficios no directos esperados son inferiores a los riesgos y molestias previstos. Las advertencias sobre la lectura de la tabla son las mismas que las señaladas en el punto 32.1 para la tabla 2. 81 en un contexto de cuidado y estima al paciente y si manifestó voluntades anticipadas 33.1 Contexto de cuidado y estima. Cuando se dan las condiciones señaladas, antes de la inclusión en un estudio clínico habría que comprobar que la persona que dará el consentimiento por representación es una persona «éticamente adecuada», es decir, juiciosa, que ha mantenido una relación de confianza con el paciente y, por lo tanto, conocía sus valores, que cuida de él y le quiere.96 Hay que alejar cualquier posibilidad de que la inclusión forme parte de una relación o situación de indiferencia o abandono. 33.2 Instrucciones previas. Cuando se dan las condiciones señaladas, antes de la inclusión en un estudio clínico debería indagarse entre otros miembros de la familia o conocidos del paciente si éste manifestó en algún momento rechazo a participar en este tipo de estudios y si lo decía sinceramente. 33.3 «Investigaciones de emergencia». Algunas veces los protocolos de investigación se diseñan para investigar patologías que se dan de una manera repentina, por ejemplo trauma cerebral, parada cardiopulmonar o accidente vascular encefálico. En estas situaciones, en las cuales no es posible obtener el consentimiento de la persona afectada y no se dispone de tiempo suficiente para localizar a una persona que pueda autorizar la participación en el estudio clínico, se hace evidente la necesidad de promover el documento de instrucciones previas y que los profesionales sanitarios tengan rápido acceso a él. Sobre las investigaciones de emergencia que pueden ser anticipadas, el comentario sobre la pauta 6 de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002) dice lo siguiente: «Si es posible, se debería intentar identificar a la población que, probablemente, desarrollará la condición que será estudiada. Eso puede hacerse fácilmente, por ejemplo, si se trata de una condición que se repite periódicamente en los individuos, como en caso de convulsiones epilépticas o embriaguez. En tales casos, se 96 La posición 4 de la Position Statement Informed Consent for Research on Human Subjects with Dementia American Geriatrics Society Ethics Committee (2007) habla de «sustitutos tradicionales» (traditional surrogates), en la mayoría de casos, familiares o personas que aman y conocen a la persona y su sistema de valores. 82 debería contactar con los potenciales sujetos mientras sean plenamente capaces de otorgar el consentimiento informado e invitarlos a participar en la investigación en futuros periodos de incapacidad». 34 Cuando los riesgos y las molestias de un tratamiento médico experimental en personas incapacitadas legalmente son muy elevados, debería disponerse de autorización judicial 34.1 Ni el artículo 6 del Convenio de Oviedo sobre Derechos Humanos y Biomedicina (1997), ni el artículo 9.3 de la Ley española 41/2002 básica reguladora de la autonomía del paciente, ni el título X del libro primero del Código civil (De la tutela, de la curatela y de la guarda de los menores e incapacitados), establecen la necesidad de obtener la autorización judicial para aplicar a una persona incapacitada tratamientos médicos que puedan poner en peligro su vida o su integridad física o psíquica. Los únicos supuestos que contempla la ley española sobre la necesidad de disponer de la autorización judicial son la esterilización de personas incapacitadas (art. 428 del Código penal) y el ingreso en un centro psiquiátrico (art. 763 de la Ley de Enjuiciamiento Civil). El artículo 219.1 del Código de familia de Cataluña sí requiere la autorización judicial en estos casos, y dice así: «La persona titular de la tutela necesita autorización judicial para: […] (b) Aplicar a la persona incapacitada tratamientos médicos que fundamentalmente puedan poner en grave peligro su vida o su integridad física o psíquica.» 35 Cuando el paciente no es plenamente competente y no ha sido incapacitado legalmente, debe obtenerse también la autorización de las personas que le cuidan, aunque eso hoy no esté previsto legalmente 35.1 La decisión participada o compartida. La ley prevé situaciones en las cuales sea necesario tomar una decisión, el paciente no esté en condiciones de hacerlo y no haya representante legal, ante lo cual «el consentimiento lo prestarán las personas vinculadas a él por razones familiares o de hecho.»97 Esta posibilidad, sin embargo, parece que la ley la prevé sólo para casos de urgencia. 97 Art. 9.3.a de la Ley española 41/2002. Art. 7.4 del RD 223/2004. Art. 10.5 del Código de ética y deontología del Consejo General de Colegios Médicos de España. Art. 75 del Código de deontología del Consejo de Colegios de Médicos de Cataluña. Art. 28 de la Declaración de Helsinki (2008). 83 Cuando el paciente no tiene plena capacidad de comprensión y decisión para participar en un estudio clínico y no está incapacitado legalmente, la decisión debe extenderse al círculo de personas que le cuidan. Una decisión participada o compartida es una medida prudente que implica informar a la familia de una decisión que también les afecta como personas cuidadoras, lo cual permite que la persona sin plena capacidad reciba el apoyo necesario para tomar una decisión. Sin embargo, este procedimiento podría suponer la vulneración del derecho a la intimidad del paciente que no ha sido incapacitado legalmente. Por este motivo, en estos casos, y siempre que sea posible, debe pedirse autorización al paciente para informar a sus familiares o a las personas que le cuidan. En este sentido, el artículo 5.1 de la Ley española 41/2002 dice: «El titular del derecho a la información es el paciente.También serán informadas las personas vinculadas a él, por razones familiares o de hecho, en la medida que el paciente lo permita de manera expresa o tácita». Si el paciente sin plena capacidad para decidir y sin haber sido incapacitado legalmente no autoriza compartir esta información con su familia o con las personas que le cuidan, no podrá ser incluido en el estudio clínico. 35.2 La decisión participada o compartida debería incorporarse al cuerpo legislativo. Actualmente la decisión participada o compartida ya se considera una buena práctica clínica en las situaciones en las cuales el paciente no tiene plena capacidad para decidir y no está incapacitado legalmente. Por este motivo, en las hojas de consentimiento de las investigaciones clínicas dirigidas a estas personas figura también, junto al espacio para la firma del paciente, otro para la firma de la persona que le cuida. Ahora bien, esta práctica no está prevista legalmente, porque jurídicamente la capacidad para tomar una decisión de este tipo es categórica: una persona es competente o no lo es para hacerlo. Consideramos necesario incorporar en el cuerpo legislativo la decisión participada, o figuras como el asistente o acompañante para decisiones concretas, lo cual posibilitaría no sólo la actual representación en la toma de decisiones, sino también el apoyo o asistencia en la toma de decisiones. En determinadas situaciones, el 84 apoyo en la toma de decisiones puede convertirse en una respuesta adecuada a la necesidad de protección sin tener que recurrir a la incapacitación. 36 Cuando el paciente no es plenamente competente y no ha sido incapacitado legalmente, debería advertirse que, como criterio general, prevalece la decisión del paciente. Y en los casos en que se considere que no es prudente atender su voluntad, el criterio general debería ser la intervención del juez 36.1 El criterio general es que hay que atender la voluntad de la persona afectada. La legislación vigente señala que el consentimiento por representación sólo es posible en los casos de incapacitación legal o de intervenciones indispensables.98 Por lo que se refiere a la primera excepción, si la persona no es plenamente capaz de dar el consentimiento pero no ha sido incapacitada, como criterio general debe respetarse su voluntad. Con respecto a la segunda excepción, las situaciones en las cuales un estudio clínico se convierte en una intervención indispensable desde el punto de vista clínico a favor de la salud de la persona afectada son excepcionales. El criterio general, por lo tanto, es que hay que atender la voluntad de la persona afectada. 36.2 La decisión en estudios clínicos con posibles riesgos y molestias (R-M), de los cuales se espera obtener un beneficio directo (BD) para pacientes no incapacitados legalmente pero sin plena capacidad de decisión. En estos casos y siempre que se cumplan todos los requisitos señalados por la ley y en estas recomendaciones, que deberán haber sido analizados y aprobados por un CEIC, se pueden dar las siguientes situaciones: 98 Art. 9 de la Ley española 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica. 85 TABLA 4 BD ≈ R-M BD > R-M99 BD < R-M100 Paciente no incapacitado legalmente, pero sin plena capacidad para obrar sí no sí no sí no sí no sí no sí no Persona o personas que le cuidan sí sí no no sí sí no no sí sí no no Decisión final que consideramos más prudente sí sí/ no sí no sí no sí/ no no sí no sí/ no no K L M N O P Situaciones susceptibles de profundización Situaciones L, M y O. El paciente quiere participar en un estudio con muchas posibilidades de que le sea beneficioso, y la persona o personas que le cuidan consideran que no debería hacerlo (L). El paciente no quiere participar en un estudio en el cual no es posible valorar si prevalecen los beneficios directos o los riesgos y/o molestias, y la persona o personas que le cuidan consideran que sí debería hacerlo (M). El paciente no quiere participar en un estudio en el cual los riesgos y/o las molestias previstos son superiores a los beneficios directos esperados, y la persona o personas que le cuidan consideran que sí debería hacerlo (O). Consideramos que en estas tres situaciones no hay ninguna razón aceptable que aconseje no atender la opinión del paciente. Situaciones K, N y P. El paciente no quiere participar en un estudio con muchas posibilidades de que le sea beneficioso, y la persona o personas que le cuidan consideran que debería hacerlo 99 Tal como se ha señalado en la tabla 1: BD > R-M significa que los beneficios directos esperados (BND) son superiores a los riesgos y molestias previstos (R-M); BD ≈ R-M, que los beneficios directos esperados son equiparables a los riesgos y molestias previstos, y BD < R-M, que los beneficios directos esperados son inferiores a los riesgos y molestias previstos. Las advertencias para la lectura de la tabla son las mismas que las señaladas en el punto 32.1 para la tabla 2. 100 Con las condiciones descritas en la recomendación 12. 86 (situación K); el paciente quiere participar en un estudio en el cual no es posible valorar si prevalecen los beneficios directos o los riesgos y/o molestias, y la persona o personas que le cuidan consideran que no debería hacerlo (situación N); el paciente quiere participar en un estudio en el cual los riesgos y/o las molestias previstos son superiores a los beneficios directos esperados, y la persona o personas que le cuidan consideran que no debería hacerlo (situación P). En estas tres situaciones, las razones que pueden aconsejar no aplicar el criterio de atender la decisión de la persona afectada pueden ser diversas, por ejemplo que hacerlo puede generar un perjuicio al paciente y también a terceras personas, en este caso las que cuidan y es de suponer que cuidarán de la persona afectada a lo largo de toda su vida. En estas tres situaciones sólo un juez puede romper la obligación de atender la voluntad de la persona afectada, porque sólo el poder judicial puede suspender el ejercicio del derecho a la libertad. Por lo tanto, en los casos en que la persona o personas que cuidan del paciente consideren que éste no está en condiciones de ejercer su libertad a pesar de no estar incapacitado legalmente, deberán hacer lo siguiente: i) Iniciar un proceso de incapacitación legal. ii) Si es necesario tomar una decisión rápida, pedir al juez la aplicación de medidas cautelares.101 36.3 La decisión (i) en estudios clínicos con posibles riesgos y molestias (R-M), (ii) de los cuales no se espera obtener un beneficio directo (BND), (iii) en los que participan pacientes con una patología que les ha supuesto la pérdida brusca de la capacidad de decisión y (iv) que no tienen voluntades previas sobre la investigación clínica, o que nunca han tenido plena capacidad de decisión. En estos casos y siempre que se cumplan todos los requisitos señalados por la ley y en estas recomendaciones, que habrán sido analizados y aprobados por un CEIC, se pueden dar las situaciones siguientes: 101 Art. 762 de la Ley de enjuiciamiento civil. 87 TABLA 5 BND ≈ R-M BND > R-M102 Paciente no incapacitado legalmente, pero sin plena capacidad para obrar sí Persona o personas que le cuidan sí sí no no sí sí no no Decisión final que consideramos más prudente sí no sí/ no no sí no sí/ no no Q R S T Situaciones susceptibles de profundización no sí no sí no sí BND < R-M no Estos tipos de estudio deberían ser rechazados por el CEI y, por lo tanto, no deberían llegar al paciente, tutor, curador o guardador de hecho. Situaciones Q y S. Situaciones en las cuales el paciente no quiere participar en un estudio sin beneficios directos y en cambio la persona o personas que cuidan de él consideran que sí debería hacerlo, la ética y la ley señalan que debe prevalecer la voluntad del paciente. Situaciones R y T. En la situación R, el paciente sin plenas capacidades para decidir pero sin estar incapacitado legalmente quiere participar en un estudio clínico que no le aportará ningún beneficio directo, pero que sí puede suponer beneficios para la ciencia o para futuros pacientes, en contra de la opinión de la persona o personas que le cuidan. En la situación T, el paciente quiere participar, también en contra de la opinión de la persona o personas que le cuidan, en una investigación sin beneficios directos en la cual no es posible valorar si prevalecen los beneficios o los riesgos y/o molestias. 102 Tal y como se ha señalado en la tabla 1: BND > R-M significa que los beneficios no directos esperados (BND) son superiores a los riesgos y molestias previstos; (R-M); BND ≈ R-M que los beneficios no directos esperados son equiparables a los riesgos y molestias previstos, y BND < R-M, que los beneficios no directos esperados son inferiores a los riesgos y molestias previstos. Las advertencias para la lectura de la tabla son las mismas que las señaladas en el punto 32.1 para la tabla 2. 88 En estas dos situaciones, las razones éticas que pueden aconsejar no atender la opinión de la persona afectada es que hacerlo puede generar un perjuicio al paciente y también a las personas que cuidan y es de suponer que cuidarán de él a lo largo de toda su vida. Y la respuesta, tal como se ha indicado en las situaciones K, N y P de la tabla 4 es que sólo un juez puede alterar la obligación de atender la voluntad de la persona afectada, porque sólo el poder judicial puede suspender el ejercicio de la libertad de una persona. Por lo tanto, en los casos en que la persona o personas que cuidan al paciente consideren que a pesar de ser capaz legalmente no está en condiciones de ejercer su libertad, deberán hacer lo siguiente: i) Iniciar un proceso de incapacitación legal. ii) Si hay que tomar una decisión rápida, pedir al juez la aplicación de medidas cautelares. 37 Si la persona que ha de dar el consentimiento no sabe leer, es deseable que haya un testigo imparcial (respecto al estudio clínico) en el momento de firmar el consentimiento 37.1 La hoja de consentimiento por escrito es un documento de prueba de cumplimiento del deber de informar oral y correctamente. Tal como señala la norma 4.8.9 de la Guía de buena práctica clínica CPMP/ICH/95, «si el sujeto o su representante legal no saben leer, un testigo imparcial deberá estar presente durante la información del consentimiento informado», que también deberá firmar la hoja de consentimiento. El artículo 4.1 de la Ley española 14/2007 de investigación biomédica dice: «Si el sujeto de la investigación no pudiera escribir, el consentimiento podrá ser prestado por cualquier medio admitido en derecho que permita dejar constancia de su voluntad».103 38 En la hoja de consentimiento por escrito deberían figurar, además de los 12 ítems para el paciente y de los 14 para el tutor actualmente establecidos, los riesgos a los que se somete el paciente 103 Este artículo recoge lo que señala el artículo 3.2.d de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo. 89 38.1 La hoja de consentimiento por escrito como documento probatorio. La hoja de consentimiento por escrito es el documento probatorio del hecho que se ha recibido toda la información pertinente y de una manera adecuada, y en ella se reiteran algunos de los principales aspectos de procedimiento que ya figuran en la hoja de información. Consideramos que en un documento probatorio deberían figurar también los principales riesgos (y las molestias, en caso de que sean significativas) a que se someterá el paciente, que, no cabe olvidar, es el aspecto más importante para él y lo que puede ser motivo de litigio. 38.2 Los 12 ítems y la estructura actuales de la hoja de consentimiento por escrito para personas no incapacitadas legalmente pero sin plena capacidad son los siguientes: 1) 2) 3) Título del estudio clínico. Yo (nombre y apellidos del paciente). He sido informado oralmente, sin prisas y de un modo comprensible sobre el estudio realizado por el investigador (nombre y apellidos del investigador). 4) He podido realizar las preguntas que he considerado necesarias. 5) He recibido información suficiente y satisfactoria. 6) He leído y entendido la hoja de información. 7) He tenido tiempo suficiente para considerar mi participación en el estudio. 8) Entiendo que mi participación es voluntaria. 9) Entiendo que me puedo retirar del estudio: a) Cuando quiera/queramos. b) Sin tener que dar explicaciones. c) Sin que esto repercuta en mi atención sanitaria. 10) Doy mi conformidad para participar en el estudio. 11) Firma del paciente, del investigador y del cuidador de hecho (véase la recomendación 35) o testigo (véase la recomendación 37). Debería especificarse la relación que el guardador de hecho o el testigo mantienen con el paciente (p. ej., el grado de parentesco). 12) Lugar y fecha. 90 38.3 Los 14 ítems y la estructura actuales de la hoja de consentimiento por escrito para el tutor legal son los siguientes: 1) 2) 3) 4) 5) 6) 7) 8) 9) 10) 11) 12) 13) 14) Título del estudio clínico. Yo (nombre y apellidos del tutor). En calidad de tutor de (nombre y apellidos del paciente). He/hemos sido informados oralmente, sin prisas y de un modo comprensible sobre el estudio por el investigador (nombre y apellidos del investigador). He/hemos podido realizar las preguntas que hemos considerado necesarias. He/hemos recibido información suficiente y satisfactoria. He/hemos leído y entendido la hoja de información. He/hemos tenido tiempo suficiente para considerar la participación en el estudio. Entiendo/entendemos que la participación en el estudio es voluntaria. Declaro que el paciente nunca se ha manifestado —que yo sepa— en contra de participar en estos tipos de investigación y que, por lo tanto, esta autorización representa su supuesta voluntad. Entiendo que me puedo/nos podemos retirar del estudio: a) Cuando quiera/queramos. b) Sin tener que dar explicaciones. c) Sin que esto repercuta en mi/nuestra atención sanitaria. Doy mi conformidad para participar en el estudio. Firma del tutor y del investigador. Lugar y fecha. 39 Hay que promover las instrucciones previas también en temas de investigación 39.1 La legislación vigente prevé que las personas podamos expresar en un documento las instrucciones sanitarias que deben tenerse en cuenta cuando no sea posible hacerlo personalmente y designar a la persona o personas que queremos que sean nuestros tutores en caso de incapacitación (autotutela). En el momento de realizar un estudio clínico y el paciente no tenga capacidad de decisión y exista un documento de instrucciones previas, la ley obliga a aplicarlas siempre que no sean contrarias al ordenamiento jurídico y 91 a la lex artis.104 Las instrucciones previas van adquiriendo reconocimiento principalmente en el ámbito asistencial; sin embargo, en el de la investigación su situación es, como señalan algunos autores, poco utilizada y hasta contradictoria.105 Los profesionales sanitarios y sociosanitarios deben recomendar y promover, cuando sea posible y no añada dolor al paciente, la redacción del documento de instrucciones previas a los pacientes a los que se ha detectado una enfermedad que les llevará o puede llevar a perder la capacidad deliberativa.106 Debería informárseles, con lenguaje comprensivo y sin prisas, que en el futuro podría darse la situación de que se le pidiera, a él o a su familia y tutor, participar en una investigación clínica, del proceso que se sigue en estos casos, de las garantías que se ofrecen, de las dudas éticas que pueden surgir y de la conveniencia de que redacte un documento de instrucciones previas en el que también figure: (i) su predisposición a participar o no en investigaciones clínicas, (ii) en el caso de que lo autorice, con qué condiciones; (iii) que nombre un representante, y (iv) la posibilidad o no de establecer consigo mismo un «contrato Ulises».107 En esta situación, muchas personas prefieren: (i) nombrar a una persona de su confianza que los represente, y (ii) que se tenga en cuenta lo mejor para ellos, aunque esto y dentro de unos límites pueda suponer invalidar las instrucciones previas, cuando las haya. 104 Art. 11 de la Ley española 41/2002. Art. 15.iv del Protocolo adicional al Convenio para la protección de los derechos humanos y la dignidad del ser humano respecto a las aplicaciones de la biología y la medicina, relativo a la investigación biomédica. 105 LÖTJÖNEN, S. (2006): «Medical research on patients with dementia. The role of advance directives in European legal instruments», European Journal of Health Law (septiembre 2006), 13 (3), p. 235-261. WELIE, S.P.; BERGHMANS, R.L. (2006): «Inclusion of patients with severe mental illness in clinical trials: issues and recommendations surrounding informed consent», CNS Drugs, 20 (1), p. 67-83. 106 Así lo señala, por ejemplo, la posición 3 y la justificación de la posición 2 de la Position Statement Informed Consent for Research on Human Subjects with Dementia American Geriatrics Society Ethics Committee (2007). 107 Un «contrato Ulises» es la manifestación expresa que se cumplan sus antiguas voluntades incluso en el caso que, estando incapacitado a causa de la evolución de la enfermedad, quisiera revocarlas. 92 40 No siempre es recomendable atender las instrucciones previas en lo referente a la participación en estudios clínicos 40.1 La ley señala: «No serán aplicadas las instrucciones previas contrarias al ordenamiento jurídico, a la lex artis, ni las que no se correspondan con el supuesto de hecho que el interesado haya previsto en el momento de manifestarlas. En la historia clínica del paciente quedará constancia razonada de las anotaciones relacionadas con estas previsiones.».108 Independientemente de esta norma legal, y por las razones señaladas en las recomendaciones 37, 38 y 40, a veces no es prudente ni correcto atender las instrucciones previas de una persona en lo referente a la participación en estudios clínicos. VII. SEGUIMIENTO 41 El CEIC debe establecer procedimientos para supervisar que se cumplen las condiciones y procedimientos establecidos en el protocolo del estudio clínico 41.1 Las buenas prácticas clínicas y la legislación señalan que la investigación clínica debe ser objeto de evaluación y seguimiento por parte de los CEIC, y que la periodicidad de este seguimiento debe ser proporcional al riesgo al que se exponen las personas que participan en ella.109 También «tomará las medidas que sean oportunas con el fin de comprobar que la continuidad del proyecto está justificada a la luz de los nuevos conocimientos que se alcancen a lo largo de su ejecución».110 108 Art. 11 de la Ley española 41/2002. Pauta 7 de les Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). 109 Normas 3.1.4 y 4.10 de la Guía de buena práctica clínica CPMP/ICH./95. Art. 3.2.a de la Directiva 2001/20/CE del Parlamento Europeo y del Consejo. Art. 2.g de la Ley española 14/2007. 110 Art. 25.1 de la Ley española 14/2007 de investigación biomédica. 93 42 Cuando se producen cambios en las condiciones o procedimientos de un estudio clínico y también periódicamente en estudios a largo plazo, el investigador debe pedir de nuevo el consentimiento informado a los pacientes o tutores 42.1 Cuando se producen cambios en las condiciones o procedimientos de un estudio clínico y también periódicamente en estudios a largo plazo, hay que informar al paciente, y el investigador debe pedir nuevamente el consentimiento informado.111 111 Pauta 3.3.i de las Pautas para la buena práctica clínica (BPC) en ensayos con productos farmacéuticos (OMS-1995). Norma 4.8.2 y 4.8.11 de la Guía de buena práctica clínica CPMP/ICH./95. Comentario sobre la pauta 4 («Renovación del consentimiento») de las Pautas éticas internacionales para la investigación biomédica en seres humanos (2002). El art. 25.5 de la Ley 14/2007 de investigación biomédica considera que basta con informar al paciente o a su representante legal. Dice así: «Cualquier información relevante sobre la participación en la investigación tiene que ser comunicada a los participantes o, si procede, a sus representantes, con la máxima brevedad». 95 BIBLIOGRAFÍA, DECLARACIONES Y LEGISLACIÓN ANDERSON, J.; HONNETH,A. (2004): «Autonomy, vulnerability and justice», en CHRISTMAN, J.; ANDERSON, J. (ed.): Autonomy and the challenges to liberalism, Cambridge, Cambridge University Press, p. 127-149. ANNA, G.; GRODIN, M. (1992): The Nazi Doctors and the Nuremberg Code, Oxford, Oxford University Press. APPELBAUM, P.S., LOREN, H.R.; LIDZ, CH.W.; BENSON, P.; WINSLADE, W. (1987): «False hopes and best data: consent to research and the therapeutic misconception», The Hasting Center Report, vol. 12, n.º 2, p. 20. APPELBAUM, T.L.; GRISSO T. (1986): «Assessing patient’s capacities to consent to treatment», New England Journal of Medicine (NEJM), 1988, 319 (25), p. 1635-8. – (1986): A history and theory of informed consent, New York, Oxford University Press. – (1995): «The MacArthur treatment competence study», Law and Human Behavior, 19, p. 105-174. – (1998): Assessing competence to consent to treatment: a guide for physicians and other health professionals, New York, Oxford University Press. – (2001): MacCAT-CR: MacArthur Competence Assessment Tool for Clinical Research, Sarasota (Florida), Professional Resource Press. ARRIOLA MANCHOLA, E.; IBARZÁBAL ARAMBERRI, X. (2007): «Fundamentos éticos de respeto al enfermo», en MARTÍNEZ LAGE, J.M.; CARNERO PARDO, C. (editores): Alzheimer 2007: recapitulación y perspectivas, Madrid, Laboratorios Andrómaco, 2007. BARTON, W.; PALMER, PH.D. [et al.] (2005): «Assessment of capacity to consent to research among older person with schizophrenia, Alzheimer disease, or diabetes mellitus», Archives of General Psychiatry, vol. 62, julio 2005. BEAUCHAMP, T.L.; CHILDRESS, J.F. (1994): Principles of Biomedical Ethics (Fourth Edition) (trad. castellana de Gracia, T., Júdez F.J. y Feito, L.: Principios de ética biomédica, Barcelona, Masson, 1998, p. 130-134). BEAN, G. [et al.] (1994): «The psychometric properties of the competency interview schedule», Canadian Journal of Psychiatry, 39, p. 368-376. BERTOLI, A.M. (2007): «Lack of correlation between satisfaction and knowledge in clinical trials participants: A pilot study», Contemporary Clinical Trials, 23 de mayo de 2007. BOADA ROVIRA, M.; ROBLES BAYÓN, A. (ed.) (2009): Documento Sitges 2009. Capacidad para tomar decisiones durante la evolución de una demencia: reflexiones, derechos y propuestas de evaluación, Barcelona, Editorial Glosa. BODENHEIMER, TH. (2000): «Uneasy alliance. Clinical investigators and the pharmaceutical industry», New England Journal of Medicine, vol. 342, 2000, p. 1539-1544. BOFILL, X.; URRUTIA, G.; ALONSO, P.; ROURA, M. (2007): «La participación de los pacientes en los ensayos clínicos», Humanitas. Humanidades Médicas, n.º 17, julio 2007. 96 BRODY, H. (1998): The ethics of biomedical research, Oxford, Oxford University Press. BUCHANAN, A.; BROCK, D.W. (1989): Deciding for others. The ethics of surrogate decision-making, New York, Oxford University Press. BUCHANAN, A. (2004): «Mental capacity, legal competence and consent to treatment», Journal of de Royal Society of Medicine, n.º 97, p. 415-420. BUCKLES, V.D. [et al.] (2003): «Understanding of informed consent by demented individual», Neurology, 61, p. 1662-1666. CALE, G.S. (1999): «Risk-related standards of competence: Continuing the debate over riskrelated standards of competence», Bioethics, 13, p. 131-148. CARNEY, M. [et al.] (2001): «The development and piloting of a Capacity Assessment Tool», The Journal of Clinical Ethics, 2001, 12, p. 17-23. DEMARCO, J.P. (2002): «Competence and paternalism», Bioethics, 16 (3), p. 231-245. DEPARTMENT OF HEALTH AND HUMAN SERVICES (EE.UU.). «Common Rule, 45 CFR 46. Federal Policy for the Protection of Human Subjects; Notices and Rules», Federal Register, 1991, §102 (i). DERENZO, E.G.; CONLEY, R.R; LOVE, R.C. (1998): «Assessment of capacity to gide consent to research participation: State of the art and beyond», The Journal of Health Care Law and Policy, 1, p. 66-87. DILIP, V.; JESTE, M.D. [et al.] (2007): «A new brief instrument for assessing decisional capacity for clinical research», Archives of General Psychiatry, 64 (8), p. 966-974. DILIP, V.; JESTE, M.D.; SAKS, E.S. (2006): «Decisional capacity in mental illness and substance use disorders: empirical database and policy implications», Behavioral Sciences and the Law, 24, p. 607-628. DRANE, J. (1984): «Competency to give an informed consent. A model for making clinical assessments», JAMA (The Journal of the American Medical Association), vol. 252, núm. 7, p. 925-927. (1985): «The many faces of competency», The Hastings Center Report, vol. 15, n.º 2, p. 17-21. DRAPER, R.J.; DAWSON, D. (1990): «Competence to consent to treatment: A guide for the psychiatrist», Canadian Journal of Psychiatry, 35, p. 285-289. DUNN, L. B. [et al.] (2006): «Assessing decisional capacity for clinical research or treatment: A review of instruments». American Journal of Psychiatry (agosto 2006), 163, p. 1323-1334. EDELSTEIN, B. (1999): Hopemont Capacity Assessment Interview Manual and Scoring Guide, Mongantown, W.V., West Virginia University, 1999. ELLEBERG, S.S.; TEMPLE, R. (2000): «Placebo controlled trials and active control trials in the evaluation of new treatment. II: practical issues and specific cases», Annals of Internal Medicine, 133, p. 464-470. EMANUEL, E.; STEINER, D. (1995): «Institutional conflict of interest», New England Journal of Medicine, vol. 332, 1995, p. 262-267. ETCHELLS, E. [et al.] (1999): «Assessment of patients capacity to consent to treatment», Journal of General Internal Medicine, 1999, 14, p. 27-34. 97 FITTEN, L.J.; WAITE, M.S. (1990): «Impact of medical hospitalization on treatment decisionmaking capacity in the elderly», Archives of Internal Medicine, 150, p. 1717-1721. GAUTHIER, S. [et al.]: Alzheimer’s Disease and Related Disorders (trad. castellana de E. Carreras: Enfermedad de Alzheimer y transtornos relacionados, Barcelona, Ars Medica, 2006). HONNETH, A. (1992): Kampf um Anerkennung (trad. castellana de M. Ballestero: La lucha por el reconocimiento. Por una gramática moral de los conflictos sociales, Barcelona, Crítica, 1997). JANOFSKY, J.S.; MCCARTHY, R.J.; FOLSTEIN, M.F. (1992): «The Hopkins Competency Assessment Test: A brief method for evaluating patients’ capacity to give informed consent», Hospital and Community Psychiatry, 43, p. 132-136. JOFFE, S. [et al.] (2001): «Quality of informed consent in cancer clinical trials: a cross-sectional survey», The Lancet, vol. 358, p. 1772-1777. – (2001): «Quality of informed consent: A new measure of understanding among research subjects», Journal of the National Cancer Institute, 93, p. 139-147. KANT, I. (1785): Grundlegung zur Metaphysik der Sitten (trad. castellana de R. R. Aramayo: Fundamentación para una metafísica de las costumbres, Madrid, Alianza Editorial, 2002). – (1797): Metaphysik der Sitten, II §38 (trad. castellana d’A. Cortina y J. Conill: La metafísica de las costumbres. Segunda parte. Principios metafísicos de la doctrina de la virtud, Madrid, Tecnos, 2002). KARLAWISH, J. (2004): «Ethics of research in dementia» (trad. castellana: «Ética en la investigación sobre la demencia», en GAUTHIER, S. [et al.]: Enfermedad de Alzheimer y trastornos relacionados, Barcelona, Ars Medica, 2006. KARLAWISH J.; KIM S.Y.; KNOPMAN, D.; VAN DYCK, C.H.; JAMES, B.D.; MARSON, D. (2008): «Interpreting the clinical significance of capacity scores for informed consent in Alzheimer disease clinical trials», American Journal of Geriatric Psychiatry (jul. 2008), 16 (7), p. 568-574 (Epub 12 junio 2008). KARLAWISH, J.; RUBRIGHT J.; CASARETT, D.; CARY, M.; TEN HAVE, T.; SANKAR, P. (2009): «Older adults’ attitudes toward enrollment of non-competent subjects participating in Alzheimer’s research», en American Journal of Psychiatry (febr. 2009), 166 (2), p. 182-188 (Epub 2008, 15 oct.). KIM, S.Y.H [et al.] (2009): «Surrogate consent for dementia research. A national survey of older Americans», Neurology, 2009, 72, p. 149-155. LEVINEM, R.J. (1999): «The need to revise the Declaration of Helsinki», New England Journal of Medicine, 341, p. 531-534. LÖTJÖNEN, S. (2006): «Medical research on patients with dementia. The role of advance directives in European legal instruments», European Journal of Health Law (septiembre 2006), 13 (3), p. 235-261. LUNA, F. (2001): Ensayos de bioética. Reflexiones desde el Sur, México, Fontamara. LYONS, D.J. (1999): «Use and abuse of placebo in clinical trials», Drug Information Journal, 33, p. 261-264. MACKLIN, R. (2003): «Dignity is a useless concept. It means no more than respect for persons or their autonomy», British Medical Journal, vol. 327, p. 1419-1420. 98 MARSON MCCODY [et al.] (1995): «Neuropsychological predictors of competency in Alzheimer’s disease using a rational reason legal standard», Archives of Neurology, 1995, 52, p. 955-959. MILLER, C.K. [et al.] (1996): «The Deaconess Informed Consent Comprehension Test: An assessment tool for clinical research subjects», Pharmacotherapy, 16, p. 872-878. MILLER, R.; WILLNER, H.S. (1974): «The Two-part Consent Form: a suggestion for promoting free and informed consent», New England Journal of Medicine, 290, p. 964-966. MITTAL, D.; PALMER, B.W.; DUNN, L.B.; LANDES, R.; GHORMLEY, C.; BECK, C.; GOLSHAN, S.; BLEVINS, D.; JESTE, D.V. (2007): «Comparison of two enhanced consent procedures for patients with mild Alzheimer disease or mild cognitive impairment», American Journal of Geriatric Psychiatry (febrero 2007), 15 (2), p. 163-167. MOYE, J. [et al.] (2006): «Empirical advances in the assessment of the capacity to consent to medical treatment. Clinical implications and research needs», Clinical Psycology Review, 2006, 26, p. 1054-1077. NORDENFELT, L. (2004): «The Varieties of Dignity», Health Care Analysis, 12 (2004). OLDE RIKKERT, M.G.M.; VAN DEN BERCKEN, J.H.L. (1997): «Experienced consent in geriatrics research: a new method to optimize the capacity to consent in frail elderly subjects», Journal of Medical Ethics, 1997, 23, p. 271-276. PINKER, S. (2008): «The Stupidity of Dignity. Conservative bioethics’ latest, most dangerous ploy», The New Republic (28 de mayo de 2008). ROTH, L.H. [et al.] (1982): «Competency to decide about treatment or research: An overview of some empirical data», International Journal of Law and Psychiatry, 5, p. 29-50. ROTH, L.H.; MEISEL, A.; LIDZ, C.W. (1977): «Test of Competency to Consent to Treatment», American Journal of Psychiatry, 1977, 134 (3), p. 279-286. ROTHMAN, D. (1991): Strangers at the bedside, New York, Basic Books. SACHS, G. [et al.] (1994): «Ethical aspects of dementia research: informed consent and proxy consent», Journal of Clinical Research, 42, p. 403-412. SAKS, E.R. (2002): «The California Scale of Appreciation: A new instrument to measure the appreciation component of capacity to consent to research», American Journal of Geriatric Psychiatry, 10, p. 166-174. SIMÓN LORDA, P. (2004): «El consentimiento informado: alianza y contrato, deliberación y decisión», en COUCEIRO, A. (2004): Ética en cuidados paliativos, Madrid, Triacastela, p. 79-108. – (2008): «La capacidad de los pacientes para tomar decisiones: una tarea todavía pendiente», Revista de la Asociación Española de Neuropsiquiatría, 2008, vol. XXVIII, n.º 102, p. 325-348. STANLEY, B. [et al.] (1984): «The elderly patient and informed consent: empirical findings», JAMA, 252, p. 1302-1306. STURMAN, E.D. (2005): «The capacity to consent to treatment and research: a review of standardized assessment tools», Clinical Psycology Review, 2005, 25, p. 954-974. 99 TEMPLE, R.; ELLENBERG, S.S. (2000): «Placebo controlled trials and active control trials in the evaluation of new treatment. I: ethical and scientific issues», Annals of Internal Medicine, 133, p. 455-463. TOMÀS D’AQUINO (1272?): Super Epistolas S. Pauli lectura. Ad Romanos. TOMODA, A. [et al.] (1997): «Validity and reliability of structured interview for competency incompetency assessment testing and ranking inventory», Journal of Clinical Psychology, 53, p. 443-450. TORRALBA, F. (2005): ¿Qué es la dignidad humana? Ensayo sobre Peter Singer, Hugo Tristram Engelhardt y John Harris, Barcelona, Herder, 2005. VICTOR, W.C. LUI [et al.] (2009): «Capacity to make treatment decisions in Chinese older persons with very mild dementia and mild Alzheimer disease», American Journal of Geriatric Psychiatry, 17 (5), p. 428-436. WARNER, J.; MCCARNEY, R.; GRIFFIN, M.; HILL, K.; FISHER, P. (2008): «Participation in dementia research: rates and correlates of capacity to give informed consent», Journal of Medical Ethics (marzo 2008), 34 (3), p. 167-170. WEATHERALL, D. (2000): «Academia and industry: increasingly uneasy bedfellows», The Lancet, vol. 355, 2000, p. 1574. WELIE, S.P.; BERGHMANS, R.L. (2006): «Inclusion of patients with severe mental illness in clinical trials: issues and recommendations surrounding informed consent», CNS Drugs, 20 (1), p. 67-83 WEYERS, W. (2003): The abuse of man. An illustrated history of dubious medical experimentation, New York, Ardor Scribendi, LTD. WICCLAIR, M.R. (1991): «Patient decision-making capacity and risk», Bioethics, vol. 5, n.º 2, p. 95. (1991): «A response to brock and skene», Bioethics, vol. 5, n.º 2, p. 120. WILKS, I. (1997): «The debate over risk-related standards of competence», Bioethics, 11, p. 419420. (1999): «Asymmetrical competence» Bioethics, 13, p. 154-159. WIRSHING, D.A. [et al.] (1998): «Informed consent: Assessment of comprehension», American Journal of Psychiatry, 155, p. 1508-1511. WONG, J.G. [et al.] (2000): «The capacity of people with a mental disability to make a health care desicion», Psychological Medicine, 2000, 30, p. 295-306. 1966. Pacto internacional de los derechos civiles y políticos (ICCPR).Aprobado por la Asamblea General de las Naciones Unidas en 1966 y ratificada por España en 1976. 1990. Tribunal Constitucional de España. Sentencia número 120/1990 (27 de junio de 1990). 1994. Tribunal Constitucional de España. Sentencia número 57/1994 (28 de febrero de 1994). 100 1995. Guía de buena práctica clínica CPMP/ICH/95 (Committee for Proprietary Medicinal Products/International Conference on Harmonisation) de la Agencia Europea de Medicina de la Comisión Europea. 1995. Pautas para la buena práctica clínica (BPC) en ensayos con productos farmacéuticos (OMS-1995). 1997. Convenio para la protección de los derechos humanos y la dignidad del ser humano con respecto a las aplicaciones de la Biología y la Medicina (Convenio relativo a los derechos humanos y la biomedicina). Aprobado por el Comité de Ministros del Consejo de Europa el 19 de noviembre de 1996. Abierto a la firma de los Estados el 4 de abril en Oviedo, y ratificado por las Cortes Generales Españolas el 5 de octubre de 1999. 1999. Código de ética y deontología médica del Consejo General de Colegios Médicos de España. 2000. Ley catalana 21/2000, de 29 de diciembre, sobre los derechos de información concernientes a la salud y la autonomía del paciente. 2001. Directiva 2001/20/CE del Parlamento Europeo y del Consejo, de 4 de abril de 2991, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas de los Estados miembros sobre la aplicación de buenas prácticas clínicas en la realización de ensayos clínicos de medicamentos de uso humano. 2002. Comité Consultatif National d’Éthique, aviso n.º 73 (4 de septiembre de 2002). 2002. Ley española 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica. 2002. Pautas éticas internacionales para la investigación biomédica en seres humanos. Preparadas por el Consejo de Organizaciones Internacionales de las Ciencias Médicas (CIOMS) en colaboración con la Organización Mundial de la Salud (OMS). 2004. Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos. 2005. Código de deontología del Consejo de Colegios de Médicos de Cataluña. 2005. Declaración Universal sobre Bioética y Derechos Humanos (UNESCO). 2005. Protocolo adicional al Convenio sobre los Derechos Humanos y la Biomedicina, relativo a la investigación biomédica (Consejo de Europa). 2007. Ley española 14/2007, de 3 de julio, de investigación biomédica. 2007. Position Statement Informed Consent for Research on Human Subjects with Dementia American Geriatrics Society Ethics Committee. 2007. Sentencia del Tribunal Supremo. Sala Civil. Resolución número 1216/2007, de 28 de noviembre de 2007, sobre responsabilidad médica. 2008. Declaración de Helsinki. 2009. Sentencia del Tribunal Supremo. Sala Civil. Resolución número 2/2009, de 21 de enero, sobre responsabilidad civil médica y la validez de la información oral y del consentimiento en una intervención quirúrgica. 101 PERSONAS QUE HAN ELABORADO O COLABORADO EN ESTA GUÍA Canimas Brugué, Joan (redactor y coordinador) Filósofo. Coordinador científico del Observatorio de Ética Aplicada a la Intervención Social Ayala Estrella, Esther Asociación de Familiares de Enfermos de Alzheimer de Girona Camps Rovira, Gemma Trabajadora social del Instituto de Asistencia Sanitaria (IAS) Del Pozo Àlvarez, Joan M. Filósofo. Profesor del Departamento de Filosofía de la Universidad de Girona Ferrer Beltran, Jordi Jurista. Profesor de filosofía del derecho de la Universidad de Girona Garre Olmo, Josep Psicólogo y epidemiólogo. Técnico de la Unidad de Investigación del Instituto de Asistencia Sanitaria (IAS) López-Pousa, Secundí Neurólogo. Coordinador de la Unidad de Valoración de la Memoria y las Demencias del Instituto de Asistencia Sanitaria (IAS) Monserrat Vila, Sílvia Traductora e intérprete. Secretaria técnica del Comité de Ética de Investigación Clínica del Instituto de Asistencia Sanitaria (IAS) Molinuevo Guix, José Luis Neurólogo. Coordinador de la Unidad de Alzheimer y Otros Trastornos Cognitivos del Hospital Clínic de Barcelona Pereda Gámez, Francisco Javier Magistrado y presidente de la Sección 14.ª de derecho civil de la Audiencia Provincial de Barcelona 102 Vilalta Franch, Joan Psiquiatra del Instituto de Asistencia Sanitaria (IAS) Agradecemos las observaciones de: Presas Vidal, Jordi Presidente de la Asociación de Familiares de Enfermos de Alzheimer de Cassà de la Selva (Girona) colección MATERIALES DE ÉTICA APLICADA A LA INTERVENCIÓN SOCIAL, 2 C M Aspectos éticos y jurídicos de la investigación clínica con personas adultas incapaces. Guía de análisis para los Comités Éticos de Investigación Clínica Y CM JOAN CANIMAS i BRUGUÉ (coord.) CY CMY K con el patrocinio de: con la colaboración de: Autonomía e Investigación Clínica 2010 MY colección MATERIALES DE ÉTICA APLICADA A LA INTERVENCIÓN SOCIAL, 2 observatori d’ètica aplicada a la intervenció social C A M P U S A R N A U D ’ E S C A L A Innovació i Recerca Social i Sociosanitària