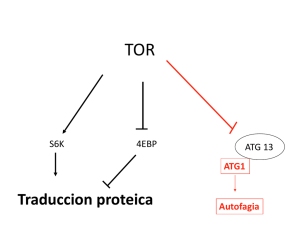
Traducido del inglés al español - www.onlinedoctranslator.com DIARIO DE HEPATOLOGÍA Revisar Autofagia en la adaptación hepática al estrés Younis Hazari1,2,3, José Manuel Bravo-San Pedro4, Claudio Hetz1,2,3,5,⇑, Lorenzo Galluzzi6,7,8,9,y, Guido Kroemer4,9,10,11,12,13,⇑,y Resumen Palabras llave: Aggrephagy; La autofagia es un proceso evolutivamente antiguo mediante el cual las células eucariotas eliminan el material citoplasmático desechable o potencialmente peligroso, para apoyar el metabolismo bioenergético y adaptarse al estrés. La evidencia acumulada indica que la autofagia opera como un mecanismo crítico de control de calidad para el mantenimiento de la homeostasis hepática en los compartimentos parenquimatosos (hepatocitos) y no parenquimatosos (células estrelladas, células endoteliales sinusoidales, células de Kupffer). De acuerdo con esta noción, la autofagia insuficiente se ha implicado etiológicamente en la patogénesis de múltiples trastornos hepáticos, incluida la deficiencia de alfa-1-antitripsina, la enfermedad de Wilson, la esteatohepatitis no alcohólica, la fibrosis hepática y el carcinoma hepatocelular. Aquí, discutimos críticamente la importancia de la autofagia funcional para la fisiología hepática, Autofagia mediada por acompañantes; Gotitas de lípidos; Lipofagia; Mitofagia; Respuesta proteica desplegada. Recibido el 11 de junio de 2019; recibido en forma revisada el 13 de agosto de 2019; aceptado el 28 de agosto de 2019 - 2019 Asociación Europea para el Estudio del Hígado. Publicado por Elsevier BV Todos los derechos reservados. Introducción La autofagia es un mecanismo celular altamente coordinado y filogenéticamente conservado que culmina con la degradación de entidades citosólicas desechables o potencialmente tóxicas dentro de orgánulos ácidos (es decir, lisosomas en células de mamíferos, la vacuola en células de levadura).1,2 El término autofagia (del griego antiguo 'aὐsόuC.Ao1', que significa' comerse a sí mismo ') fue introducido originalmente por los citólogos belgas Christian de Duve y Robert Wattiaeux en 1966 para distinguir la degradación de las entidades intracelulares de la del material extracelular (que se denominó' heterofagia ').3 Estas observaciones se basaron en más de una década de microscopía electrónica, fraccionamiento de tejidos y estudios funcionales en el hígado de rata, lo que llevó a la identificación de los lisosomas como el principal sitio de degradación del material endógeno y exógeno.4–12 Como referencia, se demostró que el tratamiento con glucagón estimula la glucogenólisis, la gluconeogénesis y activa el catabolismo de proteínas durante el mismo período.13 A día de hoy, 3 vías principales que conducen a la Se ha descrito la degradación lisosómica del material citosólico (Figura 1).2 La microautofagia implica la adquisición de pequeños sustratos autofágicos por orgánulos ácidos (en mamíferos, endosomas tardíos) tras la invaginación directa de la membrana.1 La autofagia mediada por chaperona (CMA) se basa en el reconocimiento La microautofagia endosómica (la única forma de microautofagia descrita en mamíferos) generalmente 1Instituto de Neurociencias Biomédicas degrada las proteínas citosólicas, ya sea en masa o de (BNI), Facultad de Medicina, Universidad forma selectiva.1 CMA tiene una multitud de sustratos, de de Chile, Santiago, Chile; 2Centro facto incidiendo en la regulación de una variedad de procesos relevantes para la homeostasis hepática, incluido el metabolismo bioenergético y la oncogénesis.14 La FONDAP de Gerosciencia (GERO), Salud Cerebral y Metabolismo, Santiago, Chile; 3Programa macroautofagia puede degradar entidades citoplasmáticas desechables (incluidos orgánulos enteros o partes de los mismos) de una manera bastante inespecífica (p.ej, cuando es activado por bioener- de Biología Celular y Molecular, Instituto de Ciencias Biomédicas, Universidad de Chile, Santiago, Chile; 4Equipe labellisée par la Ligue contre le cancer, Université de desafíos de getic), así como con mejoras París, Universidad de la Sorbona, selectividadp.ej, cuando es impulsado por daño de orgánulos).15 Se han introducido múltiples neologismos inducido para referir para específico variantes de INSERM U1138, Centre de Recherche des Cordeliers, París, Francia; 5Instituto Buck de Investigación sobre el Envejecimiento, Novato, CA, EE. UU.; macroautofagia, que incluye (pero no se limita a): Departamento de Oncología Radiológica mitofagia (carga: mitocondrias), agregofagia ogy, Weill Cornell Medical College, Nueva York, NY, EE. UU.; (carga: agregados de proteínas), pexofagia (carga: 7Sandra and Edward Meyer Cancer peroxisomas), reticulofagia (carga: retículo Center, Nueva York, NY, EUA; endoplásmico), lipofagia (carga: gotitas de lípidos), 8Departamento de Dermatología, cophagy (cargo: glucógeno) y xenofagia Escuela de Medicina de Yale, Nueva 6 (carga: patógenos citoplasmáticos).1,16 Macroau- Haven, CT, Estados Unidos; 9Université Paris Descartes / Paris La tofagia (de aquí en adelante denominada V, Paris, Francia; autofagia) ha estado involucrada en la regulación de 10Plataformas de metabolómica y biología celular, Gustave Roussy Commúltiples funciones celulares con patoImplicaciones fisiológicas para varios órganos. Instituto del Cáncer prehensive, Villejuif, Francia; incluido el hígado.17,18 11Pôle de Biologie, Hôpital Européen La evidencia acumulada indica que las respuestas Georges Pompidou, AP-HP, París, de proteínas solubles con un motivo KFERQ por parte de la autofágicas competentes tanto en los hepatocitos como en las familia de proteínas de choque térmico A (Hsp70) miembro células no parenquimatosas (células estrelladas, células 8 (HSPA8), junto con la translocación de dichos sustratos endoteliales sinusoidales, células de Kupffer) son clave para las que llevan KFERQ a través de las membranas lisosomales funciones fisiológicas del hígado.19 De acuerdo con esta noción, mediante una isoforma de empalme específica de proteína los defectos en la degradación del sustrato autofágico de membrana asociada a lisosoma 2 (LAMP2A).14 contribuir a la patología de una variedad de Hospital Universitario, Estocolmo, Suecia Trastornos hepáticos que incluyen deficiencia de y Comparta la coautoría senior. alfa-1-antitripsina, enfermedad de Wilson, esteatohepatitis (NASH), fibrosis hepática y hepatocelular Finalmente, la macroautofagia (Figura 2) se basa en la generación de un orgánulo de doble membrana (el autofagosoma) que secuestra material citoplasmático para su degradación y entrega esta carga a los lisosomas para su degradación.2 carcinoma (HCC).20 Aquí, discutimos el papel de la autofagia en la salud y la enfermedad del hígado. Revista de Hepatología 2020 vol. 72j 183–196 Francia; 12Instituto de Medicina de Sistemas de Suzhou, Academia China de Ciencias, Suzhou, China; 13Departamento de Salud de la Mujer y el Niño, Karolinska Revisar Autofagia en fisiología hepática compartimento de urgencias normal.38 Los desencadenantes La autofagia ocupa un lugar clave en la regulación de múltiples funciones hepáticas, así como en la preservación de la homeostasis hepática.19 reflejando al menos en parte las características biológicas únicas de este órgano (Fig. 3). En particular, los hepatocitos tienen un recambio limitado, con una vida media estimada de 6 a 12 meses, lo que los hace propensos a acumular subproductos celulares potencialmente perjudiciales en ausencia de una autofagia competente.21 Aunque los hepatocitos generalmente están inactivos, pueden reanudar rápidamente la proliferación en respuesta a una lesión, que (especialmente en presencia de defectos de autofagia) crea un riesgo de transformación maligna. 22 De manera similar, el hígado interviene en funciones centrales en el metabolismo sistémico de la glucosa y los lípidos, los cuales están íntimamente conectados con la autofagia.23,24 Además, el hígado está muy expuesto a xenobióticos y mediadores potencialmente inflamatorios de la circulación portal, y la autofagia tiene importantes efectos citoprotectores y antiinflamatorios.25,26 Finalmente, el hígado es sensible a la infección por múltiples virus hepatotrópicos, que son la causa más común de trastornos hepáticos en todo el mundo. En este contexto, las respuestas xenofágicas competentes constituyen una primera línea de defensa clave contra la infección productiva.26 Dicho esto, es importante tener en cuenta que varios virus, incluido el virus de la hepatitis B, han desarrollado estrategias para secuestrar la maquinaria autofágica para apoyar la replicación y diseminación.27,28 adicionales de la reticulofagia incluyen la privación de nutrientes El flujo autofágico basal del hígado es bastante elevado en comparación con el de otros órganos como el cerebro.19 Además, la autofagia hepática fluctúa con el comportamiento regular de ayuno de alimentación de una manera circadiana y Puntos clave La autofagia inicial es fundamental para el correcto funcionamiento del hígado en condiciones fisiológicas. puede estar fuertemente regulada por episodios de ayuno prolongados.29 Por lo tanto, en hígados de rata perfundidos, la y la infección por patógenos.38 En la reticulofagia, el reconocimiento de carga selectiva se basa en gran medida en receptores específicos que conectan las proteínas ER con la maquinaria autofágica general (Figura 2).39 Cuatro proteínas residentes en ER contienen al menos una región de interacción LC3 (LIR) que permite tales interacciones, a saber, regulador de reticulofagia 1 (RETREG1), reticulona 3 (RTN3), homólogo de SEC62, factor de translocación de preproteínas (SEC62) y progresión del ciclo celular 1 (CCPG1) (Figura 4A).40–43 RETREG1 y RTN3 también poseer a así llamado retícula homología dominio '(RHD), que favorece la curvatura y fragmentación de la membrana ER en el curso de la reticulofagia.44 Los hepatocitos generan respuestas reticulofágicas competentes al ácido oleico,45 un inductor de la enfermedad del hígado graso no alcohólico (NAFLD),46 así como al 1,4-bis [2- (3,5-dicloropiridiloxi)] benceno (TCPOBOP) y fenobarbital, ambos agonistas del receptor nuclear subfamilia 1 del grupo I miembro 3 (NR1I3) que impulsan la proliferación de hepatocitos.47 Además, se ha demostrado recientemente que RETREG1 coopera con la chaperona calnexina ER (CANX) en una respuesta reticulofágica que asegura la calidad del procolágeno.48 En particular, RETREG1 interactúa con CANX unido al procolágeno desplegado en el lumen ER, dirigiendo en última instancia tales carga resistente al proteasoma a la reticulofagia por degradación.48 Por lo tanto, la reticulofagia se destaca como un importante supresor de la secreción aberrante de colágeno (y, por lo tanto, de la fibrosis) (Figura 4B). En resumen, la reticulofagia basal e inducida por estrés contribuye a la homeostasis y adaptación hepáticas al garantizar la funcionalidad óptima del RE en los hepatocitos. proteólisis dependiente de la autofagia puede aumentar desde un nivel basal de ~ 1,5% de proteínas hepáticas totales / hora a ~ Lipofagia 4,5% de las proteínas hepáticas totales / hora en caso de El hígado es el principal órgano del metabolismo de los lípidos, inanición, lo que conduce a una pérdida de proteínas de casi asegurando no solo la transformación de primera línea de la 40%, en caso de inanición. se mantiene durante 48 horas.30,31 grasa de la dieta de la circulación portal, sino también la Se han obtenido resultados similares en hepatocitos conversión de los lípidos que son liberados en el torrente primarios cultivados.32 Estos estudios fueron los primeros sanguíneo por el tejido adiposo.49 Los hepatocitos absorben los en establecer la importancia de la autofagia para las ácidos grasos (AG) circulantes y los esterifican rápidamente en el funciones hepáticas en el contexto de la conducta de RE para producir triglicéridos y ésteres de colesterol. Tras la alimentación circadiana. La investigación posterior reveló acumulación dentro de la bicapa del RE, los lípidos neutros que múltiples variantes de autofagia específicas de la carga forman microdominios similares a lentes que son estabilizados Celular y Molecular, Segundo Piso, 33 son por proteínas como la biogénesis de gotitas lipídicas BSCL2 Sector B, Instituto de Ciencias hepática, como se analiza a continuación. ⇑ Autores correspondientes. Direcciones: Programa de Biología Biomédicas, Universidad de Chile, clave para el mantenimiento de la homeostasis inductora de almacenamiento de grasa 2 (FITM2).50 Estos Independencia 1027, Santiago, PO BOX 70086, Chile. Tel .: + 56-2-2978-6506 (C. Hetz); o Centre de Recherche des Cordeliers, Team 11, asociada, seipina (BSCL2) y la proteína transmembrana microdominios de lípidos neutros con forma de lente son Reticulofagia inestables y pueden desprenderse de la bicapa del RE debido a La forma y el volumen del retículo endoplásmico (RE), el la fluctuación térmica, lo que da como resultado la formación de principal compartimento subcelular implicado en el gotitas esféricas de lípidos (LD), que se encuentran en el plegamiento de proteínas, la secreción y la biosíntesis de citoplasma y son sitios clave para el metabolismo de los lípidos lípidos, son muy dinámicos y responden a las condiciones hepáticos (Figura 5A).50 Los LD tanto nacientes como maduros de estrés, lo que a menudo resulta en un aumento de la (generalmente 250-500 nm) están equipados con todas las (C. Hetz), kroemer @ naranja. fr(G. biogénesis.34,35 Una vez que se resuelve la homeostasis enzimas requeridas para la síntesis de triglicéridos, y su tamaño, Kroemer). reticular, las células eliminan el exceso de RE para recuperar interacción física y diafonía funcional con otros orgánulos son funciones fisiológicas a través de la reticulofagia,36,37 todos finamente regulados. 15 rue de l'Ecole de Médecine, 75005 París, Francia. Tel .: + 33-1-4427- 7667 (G. Kroemer). Correos electrónicos: chetz @ med. uchile.cl, chetz @ buckinstitute. org que también opera en la línea de base para preservar un 184 Revista de Hepatología 2020 vol. 72j 183–196 DIARIO DE HEPATOLOGÍA lated. Por lo tanto, el agotamiento de la atlastina GTPasa 1 Autofágico sustratos (ATL1), una GTPasa involucrada en la remodelación del RE, o Microautofagia la proteína accesoria del receptor 1 (REEP1), una proteína clave para la generación de túbulos del RE, afecta drásticamente el tamaño de las LD.51,52 Además, el complejo Autofágico sustratos de la proteína de coatómero 1 (COPI) regula la tensión superficial de las LD eliminando los fosfolípidos, de facto favoreciendo su fusión con las membranas del RE.53 Los Lisosoma KFER coatómeros COPI también son importantes para el suministro del dominio de fosfolipasa tipo patatina de lipasa citosólica que contiene 2 (PNPLA2) a LD.54 Sobre la privación de nutrientes, múltiples tipos de Autofagosoma LAMP2A células dan prioridad a la oxidación de ácidos grasos (FAO, también conocido como B-oxidación) como fuente de Q HSPA8 Chaperón mediado autofagia energía.55 La FAO implica la absorción mitocondrial de ácidos grasos libres para generar acetil-CoA localmente y, a través del ciclo de Krebs, NADH.+ moléculas en apoyo de la fosforilación oxidativa.56 En este escenario, las células pueden metabolizar los LD para formar AG a través de dos vías no excluyentes entre sí: i) la lipólisis, mediante la cual la proteína quinasa A (PKA) impulsa la degradación proteasomal dependiente de la fosforilación de la perilipina 1 (PLIN1), lo que a su vez permite la activación de PNPLA2; y ii) lipofagia (Figura 5A).57 La liberación de AG a partir de LD en el curso de la privación de nutrientes no solo es necesaria para que los hepatocitos sobrevivan en Macroautofagia Fig. 1. Principales variantes de autofagia en células de mamíferos. Las células de mamíferos exhiben 3 formas principales de autofagia, que pueden discriminarse entre sí en función de i) la selección del sustrato y ii) el mecanismo de entrega de carga a los lisosomas. La microautofagia es una respuesta autofágica independiente de LAMP2A que implica la invaginación directa de la membrana en la superficie de los compartimentos ácidos (lisosomas o endosomas tardíos). La autofagia mediada por chaperona depende de la translocación dependiente de LAMP2A de proteínas portadoras de KFERQ acompañadas por HSPA8 en el lisosoma. La macroautofagia se basa en la absorción de sustratos para su degradación por los autofagosomas, seguida de la fusión con lisosomas para la entrega de la carga. HSPA8, miembro 8 de la familia de proteínas de choque térmico A (Hsp70); LAMP2A, proteína 2 de membrana asociada a lisosomas, isoforma A de empalme. condiciones de escasa disponibilidad de glucosa, sino que también es fundamental para la liberación de AG y cuerpos cetónicos en el torrente sanguíneo, que sirven como sustratos energéticos en otros órganos. .58 lipofagia69 Es de destacar que la lipólisis hepática óptima se basa en el Ca operado en la tienda2+ entrada (SOCE) En consonancia con un papel central de la lipofagia en la movilización de lípidos hepáticos, el inhibidor de la regulada por la molécula de interacción estromal 1 (STIM1) y el módulo de calcio activado por liberación de calcio ORAI autofagia relativamente inespecífico 3-metiladenina (3-MA, ulator 1 (ORAI1).70 En consecuencia, los defectos en STIM1 que se dirige a VPS34)59 así como el silenciamiento del gen y ORAI1 (ya sea de forma experimental o como esencial de la autofagia ATG5 (Figura 2) favoreció la consecuencia de mutaciones de pérdida de función) dan acumulación de TG y LD en los hepatocitos.23 Se hicieron como resultado la acumulación de LD en los hepatocitos, un observaciones similares en el parénquima hepático de proceso que se acompaña de la activación compensatoria ratones sometidos a la deleción específica del hígado de de la lipofagia.70 Por lo tanto, la movilización hepática de LD otro gen esencial para la autofagia. Atg7.60 es un proceso altamente coordinado que involucra tanto a La glicina N-metiltransferasa (GNMT) se destaca como un importante regulador de la lipofagia hepática, lo que refleja su capacidad para limitar la metionina y Sadenosil-L-niveles de metionina (SAM) y, por lo tanto, previenen la activación inhibidora de la autofagia de la proteína fosfatasa 2A (PP2A).61 Por lo tanto, Gnmt- / - los hígados muestran un flujo autofágico alterado, que puede ser restaurado por inhibidores farmacológicos de PP2A.61 La lipofagia hepática óptima también se ha relacionado con el Ca2+-impulsada, caja de horquilla O1 (FOXO1) -expresión dependiente de lipasa A, tipo de ácido lisosómico (LIPA),62,63 a la actividad de pequeñas GTPasas implicadas en el tráfico vesicular, incluidos RAB7, RAB10 y RAB18,64–67 así como a la función del factor de transcripción EB (TFEB), un transactivador maestro de genes implicados en la autofagia y la biogénesis lisosomal (Figura 5B).68 CMA como a macroautofagia. De acuerdo con esta noción, Curiosamente, también se ha sugerido que CMA la activación farmacológica de la autofagia con cafeína impulsa la movilización de lípidos hepáticos en ratones, disminuyendo de manera efectiva el tamaño del compartimento LD junto con FAO.71 De acuerdo con esto, la cafeína alivia la hepatoesteatosis en ratones que reciben una dieta alta en grasas (HFD), un efecto que se ha atribuido en gran parte a la activación de la autofagia.71,72 Dicho esto, el café contiene otros inductores de la autofagia además de la cafeína, lo que significa que incluso el café descafeinado estimula un aumento del flujo autofágico en el hígado.73 de acuerdo con estudios preclínicos y epidemiológicos que sugieren que la ingesta de café reduce la incidencia de EHNA independientemente de su contenido de cafeína.74–76 La acumulación excesiva de LD en los hepatocitos puede generar una respuesta lipotóxica que implica la muerte celular dependiente de lisosomas.77 Experimentalmente, este fenotipo desempeña un papel importante en este entorno, lo que puede establecerse mediante la deleción específica de refleja el hecho de que tanto PLIN2 como PLIN3 poseen un hepatocitos de Lrp1 (que codifica la proteína relacionada con el motivo KFERQ e interactúan con HSPA8 durante la inanición. receptor de LDL 1) combinada con la suplementación con FA.78,79 69 En Por lo tanto, en comparación con sus contrapartes de control, este caso, sin embargo, la degradación dependiente de CMA de PLIN2 y PLIN3 favorece la lipólisis, no Lrp1- / - los hepatocitos son más sensibles a Revista de Hepatología 2020 vol. 72j 183–196 185 Revisar el sistema ubiquitina-proteasoma participa parcialmente en acampar ATP BCL2 VPS34 AMPK complejo PAGULK1 ATG13 AMBRA1 PAG AKT1S1 DEPTOR RPTOR MTORC1 ATG9 VPS34 su eliminación adecuada.82 En mitofagia, la selectividad se VPS15 logra a través de 3 clases diferentes de receptores de Prefagóforo 1. Iniciación 2. Nucleación Autofagosoma ATG16L1 ATG12 ATG5 ATG4 Lisosoma EDUCACIÓN FÍSICA LC3 II Podría decirse que la variante mejor caracterizada de la mitofagia es impulsada por la despolarización mitocondrial Autolisosoma y se inicia por el consiguiente bloqueo de la importación de proteínas dependiente del complejo TOMM.85,86 Carga 3. Expansión 5. Degradación Fig. 2. Regulación central de la macroautofagia en mamíferos. En células de mamíferos, las respuestas autofágicas inespecíficas (como las iniciadas por la privación de nutrientes) se pueden subdividir en 5 etapas principales: i) iniciación, ii) nucleación, iii) expansión, iv) fusión yv) degradación.Iniciación. Esta fase implica la detección bioquímica de signos de estrés bioenergético causado por la disminución de los niveles de nutrientes, incluido (pero no limitado a) el agotamiento de ATP junto con niveles aumentados de AMP cíclico. Las relaciones de AMPK / ATP altas activan AMPK y, por lo tanto, conducen: i) la fosforilación inactivante dependiente de AMPK de MTORC1; y ii) la fosforilación de activación directa dependiente de AMPK o la desfosforilación de activación indirecta (corriente abajo de la inhibición de MTORC1) de múltiples componentes del aparato de iniciación, tales como ATG13 y ULK1, y la maquinaria de nucleación, incluyendo ATG14, AMBRA1, BECN1 y UVRAG.Nucleación. En este contexto, ULK1 adquiere actividad catalítica en el contexto de un complejo supramolecular que contiene ATG13, ATG101 y RB1CC1 (mejor conocido como FIP200). Los eventos de fosforilación / desfosforilación relacionados con ULK1, AMPK y MTORC1 impulsan la nucleación de precursores de autofagosomas (también conocidos como fagophores) en el ER, aguas abajo de la síntesis de PI3P por una actividad de PI3K multiproteína de clase III que consiste en PIK3C3 (mejor conocido como VPS34), PI3KR4 (mejor conocido como VPS15), BECN1, AMBRA1 y / o UVRAG, vinculado al reclutamiento de ATG9 vesicular. El complejo VPS34 es inhibido constitutivamente por BCL2, lo que refleja la capacidad de este último para unirse físicamente e inhibir BECN1.Alargamiento. El alargamiento del fagophore está mediado por dos sistemas de conjugación de tipo ubiquitina. Por un lado, ATG7 y ATG10 impulsan secuencialmente la formación de complejos ATG12-ATG5: ATG16L1. Por otro lado, ATG4, ATG7 y ATG3 permiten la escisión de MAP1LC3B (mejor conocido como LC3) y otros miembros de la familia LC3, incluido GABARAPL1, seguido de la conjugación a PE y el reclutamiento para formar autofagosomas. LC3, GABARAPL1 y otros homólogos de LC3 proporcionan a los autofagosomas la capacidad de unirse a receptores de autofagia que contienen LIR, así como a proteínas que median la selectividad de carga, como SQSTM1 (mejor conocido como p62).Fusión y degradación. Los autofagosomas cerrados se fusionan con los lisosomas para generar autolisosomas, culminando con la acidificación luminal y la activación de hidrolasas que catalizan la degradación de la carga.2 AMBRA1, autofagia y regulador 1 de beclin 1; AMPK, proteína quinasa activada con 5 'AMP; ATG, relacionado con la autofagia; BCL2, regulador de apoptosis BCL2; BECN1, beclin 1; GABARAPL1, proteína asociada al receptor GABA tipo A como 1; MAP1LC3B, proteína 1 de cadena ligera 3 beta asociada a microtúbulos; MTORC1, diana mecanicista del complejo 1 de rapamicina; P, fosfato inorgánico; PE, fosfatidiletanolamina; PI3P, fosfatidilinositol 3-fosfato; PIK3C3, subunidad catalítica de fosfatidilinositol 3-quinasa tipo 3; PI3KR4, subunidad 4 reguladora de fosfoinositido-3-quinasa; RB1CC1, RB1 bobina en espiral inducible 1; SQSTM1, sequestome 1; ULK1, quinasa 1 de activación de autofagia similar a unc-51; UVRAG, resistencia a la radiación UV asociada. Muerte celular impulsada por FA, una respuesta citotóxica que se acompaña de niveles aumentados de SQSTM1 pero lipidación de LC3 inalterada (Figura 2), lo que refleja un bloqueo en el flujo autofágico.78,79 Se han hecho observaciones similares en el hígado de Sod1- / - ratones, que carecen de una superóxido dismutasa involucrada en las defensas antioxidantes y acumulan LD como consecuencia de una lipofagia alterada.80,81 En conjunto, estas observaciones destacan el papel clave de la lipofagia y la CMA en la regulación de la movilización de lípidos hepáticos. Esto evita el procesamiento normal y la degradación intramitocondrial de la quinasa 1 inducida por PTEN (PINK1), lo que resulta en su acumulación en el OMM y autofosforilación.87,88 Tras la fosforilación de ubiquitina y otras proteínas, incluida la mitofusina 2 (MFN2),89,90 PINK1 facilita el reclutamiento de la proteína ligasa de ubiquitina RBR E3 de parkina (PRKN), que culmina en la ubiquitinación de múltiples proteínas OMM que sirven como receptores para SQSTM1.87 Como alternativa, la maquinaria autofágica puede reclutarse en las mitocondrias dañadas por proteínas OMM no ubiquitinadas que contienen LIR, incluidas (pero potencialmente no limitadas a) proteínas que contienen BH3 como la proteína 3 que interactúa con BCL2 (BNIP3), proteína 3 que interactúa con BCL2 como (BNIP3L), BCL2 como 13 (BCL2L13) y el dominio FUN14 que contiene 1 (FUNDC1).91 Finalmente, la cardiolipina lipídica de la membrana mitocondrial interna (IMM) puede translocarse a la OMM en el contexto de daño mitocondrial, donde puede interactuar físicamente con autofagosomas nacientes a través de LC3 lipidada ( Figura 6A).92 Durante el ayuno, los hepatocitos movilizan lípidos para el resto del organismo mediante lipólisis y lipofagia (como se describió anteriormente), un proceso que se maximiza por los rápido autofágico degradación de de lo contrario, mitocondrias competentes de la FAO.93,94 En este escenario, la captación selectiva de mitocondrias por los autofagosomas puede representar hasta el 85% de los eventos autofágicos,93,94 y puede iniciarse tan pronto como 30 minutos después de la abstinencia de nutrientes.93,95 La mitofagia basal también es clave para el mantenimiento de la homeostasis en los hepatocitos, ya que preserva la calidad de la red mitocondrial, evitando la generación de especies reactivas de oxígeno (ROS) y la inútil hidrólisis de ATP debido a la despolarización de la membrana mitocondrial (Figura 6B).94,96,97 En resumen, la mitofagia contribuye al funcionamiento normal del hígado al prevenir la acumulación de mitocondrias dañadas, que pueden tener importantes efectos citotóxicos.98 y sintonizando el metabolismo mitocondrial para satisfacer las necesidades del organismo. Pexofagia La biogénesis y degradación del peroxisoma hepático son Mitofagia El daño mitocondrial inicia una forma de autofagia específica de la carga que elimina las mitocondrias con membranas despolarizadas.82 De hecho, aunque 186 SQSTM1; ii) proteínas no ubiquitinadas de la membrana 4. Fusión ATG3 LC3 I superficie mitocondrial sea propensa al reconocimiento por mitocondrial externa (OMM); y iii) receptores de lípidos.84 SQSTM1 ATG7 Pre-LC3 autofagia: i) receptores ubiquitinados, que hacen que la ATG12 ATG5 ATG10 la eliminación de las mitocondrias dañadas,83 estos orgánulos grandes requieren degradación lisosomal para UVRAG ATG14 MTOR MLST8 BECN1 procesos altamente dinámicos, como sugiere la vida media estimada de las proteínas peroxisomales del hígado de rata (1,5 días).99 La pexofagia es particularmente Revista de Hepatología 2020 vol. 72j 183–196 DIARIO DE HEPATOLOGÍA A C Autofagia Estado de reposo Daño Proliferación B D Autofagia Saludable hepatocito Orgánulos dañados y agregados proteicos Autofagia mi vena Saludable hepatocito escombros Autofagia Infección Antígenos Portal Limpieza de Antiinflamatorio Hiperactivo respuesta Respuestas inmunes ATP Autofagia Metabólico Metabólico demandas respuesta Portal F vena Infección Inmune respuesta Autofagia Virus Viral infección propagación Saludable hepatocito Fig. 3. Funciones clave de la autofagia en fisiología y patología hepática. (A) La autofagia ocupa un papel central en la preservación de la homeostasis hepática, porque apoya el elevado potencial regenerativo del órgano, (B) limpia los hepatocitos de subproductos potencialmente citotóxicos del metabolismo celular normal, (C) limita las respuestas inflamatorias potencialmente perjudiciales a las toxinas y antígenos de la circulación portal, (D) apoya las respuestas inmunes contra patógenos invasores y (E) juega un papel crítico en el mantenimiento del metabolismo local y sistémico. (F) Sin embargo, varios virus hepatotrópicos han adquirido la capacidad de aprovechar la maquinaria autofágica para su propio beneficio. A Autofagosoma SEC62 CCPG1 B Exceso de ER Reticulofagia Reticular homeostasis TCPOBOP Fenobarbital Reticulofagia NAFLD RETREG1-CANX Reticulofagia Fibrosis Inanición LC3 Infección por patógenos RETREG1 RTN3 ER Ácido oleico norte Fig. 4. Reticulofagia en el hígado. (A) La reticulofagia está habilitada por múltiples receptores que interactúan directamente con LC3 en la formación de autofagosomas, que incluyen RETREG1, RTN3, SEC62 y CCPG1. (B) La reticulofagia asegura la preservación de la homeostasis hepática al preservar el tamaño y la funcionalidad del RE en la recuperación de la inanición o la infección viral. Además, la reticulofagia inhibe la NAFLD inducida por ácido oleico, fenobarbital y TCPOBOP. Finalmente, el receptor de reticulofagia RETREG1 regula negativamente la secreción aberrante de colágeno (y por lo tanto la fibrosis) en interacciones funcionales con la chaperona CANX del ER. CANX, calnexina; CCPG1, progresión del ciclo celular 1; RE, retículo endoplásmico; N, núcleo; NAFLD, enfermedad del hígado graso no alcohólico; RETREG1, regulador de reticulofagia 1; RTN3, reticulón 3; SEC62, homólogo de SEC62, factor de translocación de preproteína; TCPOBOP, 1,4-bis [2- (3, sensibles a la acumulación de ROS, que se ha informado que inician la eliminación autofágica de peroxisomas tras la activación de un grupo extranuclear de ATM serina / treonina quinasa (ATM).100 En este contexto, ATM fosforila el factor de biogénesis peroxisomal 5 (PEX5), lo que favorece la ubiquitinación de PEX5 por PEX2 y conduce al reclutamiento mento de autofagosomas a través de SQSTM1 o NBR1 ( Figura 7A).100-102 Es de destacar que PEX5 también parece inhibir la autofagia al afectar el estado de fosforilación de MTORC1 (Figura 2) y la activación de TFEB (Figura 7A).103 El tamaño del peroxisoEl compartimento del mal también está regulado por la Puntos clave Las respuestas autofágicas inducidas por el estrés previenen la acumulación de potencial material inicialmente patógeno en los hepatocitos. disponibilidad de oxígeno. En particular, dominio PAS endotelial Revista de Hepatología 2020 vol. 72j 183–196 187 Revisar A B Hepatocitos ATG5KD ATG7- / - inhibición LD ER norte 3MA Autofagia Mismo GNMT FA FITM2 BSCL2 LIPA Lipofagia FA LD norte ATP RAB7 / 8/18 reguladores FA Rápido FFeaesdting PP2A Reunió Lipofagia TFEB Autofagia Lipofagia Hepatosteatosis Lipofagia Hepatosteatosis Cafeína inductores Fig. 5. La lipofagia en la salud y enfermedad hepática. (A) Los hepatocitos alimentados acumulan AG en LD que se estabilizan en asociación con el RE por proteínas que incluyen BSCL2 y FITM2. Durante el ayuno, los LD son movilizados por la lipofagia para apoyar el metabolismo tanto hepático como sistémico. (B) GNMT inhibe la hepatosteatosis, ya que promueve la lipofagia al limitar los niveles de metionina y SAM, evitando así la activación inhibidora de la autofagia de PP2A. Corroborando aún más un papel beneficioso de la lipofagia, la inhibición de la autofagia por la administración de 3-MA, así como por la regulación a la baja deATG5 o ATG7, promueve la hepatoesteatosis en entornos experimentales. 3-MA, 3-metiladenina; ATG, relacionado con la autofagia; BSCL2, biogénesis de gotitas lipídicas BSCL2 asociada, seipina; RE, retículo endoplásmico; FA, ácido graso; FITM2, proteína transmembrana inductora del almacenamiento de grasa 2; GNMT, glicina N-metiltransferasa; N, núcleo; PP2A, proteína fosfatasa 2A; SAM,S-adenosil-L-metionina; TFEB, factor de transcripción EB. A Mitofagia dependiente de PRKN Mitofagia independiente de PRKN B Hepatocitos Autofagosoma SQSTM1 Ub PRKN FUNDC1 MFN2 LC3 Citotóxico CL ROSA1 Δψ Mitocondrias dañadas (DM) Δψ Mitocondrias saludables (HM) metro Mitofagia DM BNIP3 Lipólisis Mitofagia FA FA efectos HM norte Alimentación metro Faassttiningg F Fig. 6. Papel de la mitofagia en la preservación de la homeostasis hepática. (A) DM exhibe un potencial transmembrana mitocondrial reducido (Dwmetro), permitiendo la acumulación de PINK1 en su superficie y la autofosforilación de PINK1, culminando con el reclutamiento de PRKN. PRKN cataliza la ubiquitinación de múltiples proteínas mitocondriales, incluida MFN2, que sirven como receptores que reconocen los autofagosomas en crecimiento a través de SQSTM1 y LC3. Alternativamente, la DM puede ser reconocida por LC3 a través de BNIP3, FUNDC1 y la CL lipídica restringida por la membrana mitocondrial interna. (B) En condiciones de alimentación, la mitofagia sirve en gran medida como un mecanismo de control de calidad para la DM. Sin embargo, durante el ayuno, la mitofagia también degrada la HM competente de la FAO para impulsar la liberación de AG movilizados por la lipólisis y la lipofagia. BNIP3, proteína 3 que interactúa con BCL2; CL, cardiolipina; DM, mitocondrias dañadas; FAO, oxidación de ácidos grasos; FA, ácido graso; FUNDC1, dominio FUN14 que contiene 1; HM, mitocondrias sanas; MFN2, mitofusina 2; ROSA1, Quinasa 1 inducida por PTEN; PRKN, proteína ligasa de ubiquitina parkina RBR E3; SQSTM1, secuestosoma 1. Proteína 1 (EPAS1, una proteína sensible al oxígeno mejor conocida como HIF-2a) parece impulsar la pexofagia en respuesta a una baja tensión de oxígeno, lo que produce alteraciones del metabolismo de los lípidos que recuerdan a los trastornos peroxisomales.104 Estos resultados identifican un vínculo insospechado entre la disponibilidad de oxígeno y las funciones peroxisomales. Los peroxisomas son orgánulos críticos para el metabolismo de los lípidos hepáticos y la síntesis de ácidos biliares, lo que apunta a un papel importante de la pexofagia en el mantenimiento de las funciones hepáticas La glucofagia juega un papel importante en la homeostasis normales. De acuerdo con esta noción, los estudios en de la glucosa hepática. De acuerdo con esta noción,Gaa- / -Stbd1 ratones conAtg7- / - Los hepatocitos revelaron que 70 a 80% - / - los de los peroxisomas hepáticos se degradan por autofagia. glucógeno en el corazón y el músculo esquelético, pero 105,106 Dicho muestran una reducción del 73% en el glucógeno lisosomal en el esto, falta una caracterización profunda de la pexofagia en el hígado. Glicofagia El glucógeno hepático, cardíaco y muscular se degrada en gran medida por la glucogenólisis, una vía catabólica iniciada con la activación de una de varias 188 variantes de glucógeno fosforilasa.107 Alternativamente, la glucosa se puede movilizar siguiendo la captación de gránulos de glucógeno por los autofagosomas y la activación del ácido glucosidasa alfa lisosomal (GAA).108 La glucofagia se basa en el dominio de unión de almidón proteico 1 (STBD1) que contiene LIR,109 y puede ser activado selectivamente por hipoxia y florizina, un inhibidor de los cotransportadores de sodio-glucosa de la familia de portadores de solutos 5 miembro 1 (SLC5A1) y SLC5A2 (Figura 7B).110,111 ratones no presentan alteraciones del metabolismo del hígado en comparación con Gaa- / - ratones.112 Estos hallazgos destacan la importancia crítica de STBD1 para la glucofagia hepática. Es de destacar que los niveles hepáticos de múltiples mediadores autofágicos, incluidos BECN1, SQSTM1 y GABARAPL1, no difieren entre los que padecen hambre.Gaa- / -Stbd1- / - y Gaa- / - Revista de Hepatología 2020 vol. 72j 183–196 DIARIO DE HEPATOLOGÍA ratones,109 lo que sugiere que la ausencia de STBD1 impone un defecto selectivo en la glucofagia en lugar de afectar las respuestas autofágicas en general. A B PEX5 En resumen, la glucofagia se destaca como un Cajero automático ROS Ub potencialmente un objetivo para el desarrollo de nuevas intervenciones terapéuticas. PEX2 Pexofagia Autofagia en enfermedad hepática Trastornos de acumulación El miembro 1 de la familia de serpinas A (SERPINA1, mejor conocido como alfa-1 antitripsina o AAT) es una glicoproteína de fase aguda con actividad inhibidora de elastasa, que es sintetizada predominantemente por hepatocitos y alcanza una concentración sérica de 85 a 250 mg / dl en condiciones normales. individuos sanos. 113 SER-PINA1 es un gen altamente polimórfico y existen varios mutantes de importancia patogénica, incluidos los mutantes comunes Z, Si, I, S, Br y K.113 La mutación Z resulta de una sola transición G-> A en el codón 342, generando una variante AAT distorsionada (AATE342K) que forma agregados retenidos dentro del RE de los hepatocitos.113 En consecuencia, los defectos en la degradación asociada a ER (ERAD) se han relacionado con una mayor gravedad de la enfermedad en modelos de roedores de deficiencia de AAT.114,115 Desde una perspectiva clínica, la deficiencia de AAT implica manifestaciones pulmonares y hepáticas, las últimas de las cuales incluyen hepatitis, cirrosis, CHC e insuficiencia hepática.116 Los eventos celulares que subyacen a la enfermedad hepática en este escenario incluyen inflamación, esteatosis, pérdida de hepatocitos y alteraciones fibróticas.114,116 Expresión de AATE342K en Atg5- / - Las líneas celulares dan Glucógeno SQSTM1 importante regulador del metabolismo de la glucosa hepático (y por extensión sistémico), y representa Glucosa TFEB Peroxisoma LC3 Glicofagia Fig. 7. Pexofagia y glucofagia en el hígado. (A) Los ROS son los principales impulsores de la pexofagia, aguas abajo de la activación de ATM y la consiguiente fosforilación de PEX5. El PEX5 fosforilado es un objetivo para la ubiquitinación dependiente de PEX2, por lo que sirve como receptor para el reconocimiento mediante el crecimiento de autofagosomas a través de SQSTM1 y LC3. PEX5 también favorece la pexofagia al limitar la actividad inhibidora de la autofagia de MTORC1. (B) La glucofagia hepática, que desempeña un papel fundamental en la regulación del metabolismo sistémico de la glucosa, se basa en STBD1, que actúa como receptor de LC3 y GAA, lo que facilita la degradación del glucógeno en los lisosomas. Tanto la hipoxia como la inhibición de la captación de glucosa por la florizina impulsan la glucofagia en los hepatocitos. ATM, ATM serina / treonina quinasa; GAA, glucosidasa alfa, ácido; MTORC1, diana mecanicista del complejo 1 de rapamicina; PEX5, factor de biogénesis peroxisomal 5; ROS, especies reactivas de oxígeno; SQSTM1, secuestosoma 1; expansión del compartimento autofagosómico en este entorno.123 De acuerdo con esto, los hepatocitos de pacientes con enfermedad de Wilson yAtp7b- / - los ratones muestran un mayor número de autofagosomas, lo que refleja la activación de una respuesta autofágica que previene la muerte celular provocada por la acumulación de cobre.123,124 De hecho, la inhibición de la autofagia con spautin 1 aceleró la desaparición de los hepatocitos que sucumbían al cobre. Las intervenciones farmacológicas basadas en autofagia para la deficiencia de alfa-1-antitripsina han alcanzado las pruebas clínicas de fase II. la sobreexpresión (ambas potencian la autofagia) median los efectos citoprotectores.123 Por tanto, la autofagia se destaca como un mecanismo importante para que los hepatocitos conserven la homeostasis a pesar de la (sobre la degradación proteasomal) en el control de acumulación de cobre. Sin embargo, queda por investigar si depósitos aberrantes de AAT.117 De acuerdo con esta noción, los inductores de la autofagia, como la carbamazepina o la la expresión específica de hepatocitos reforzada por rapamicina, pueden emplearse convenientemente para transgenes de AATE342K en ratones de tipo salvaje da como tratar la enfermedad de Wilson. La enfermedad por almacenamiento de glucógeno tipo 1a (GSD1a) aún está el hígado.118,119 Además, la sobreexpresión específica de otro hepatocitos de TFEB da como resultado una acumulación disminución del flujo autofágico.125 GSD1a es un trastorno limitada de AAT, una apoptosis reducida y una fibrosis metabólico hereditario que afecta el almacenamiento de suprimida en ratones portadores de un agente patógeno. glucógeno como consecuencia de defectos en el complejo SER-PINA1gene.120 En líneas similares, enzimático que convierte la glucosa-6-fosfato en glucosa. es La enfermedad de Wilson es un trastorno hereditario del Puntos clave acumulación, mientras que la inanición y TFEB de AAT que apuntan a un papel principal de la autofagia inductores farmacológicos de la autofagia, incluido el inhibidor de MTORC1, rapamicina y carbamazepina (cuyo mecanismo de acción se debate)59 fibrosis hepática fuertemente reducida en modelos de roedores de deficiencia de AAT,121 proporcionando la base para las pruebas clínicas de fase II en curso.20 GAA STBD1 MTORC1 como resultado una acumulación exacerbada de agregados resultado la activación de fuertes respuestas autofágicas en Hipoxia Florizina hepático trastorno con asociado decir, glucosa-6-fosfatasa a.125 Estas alteraciones deterioran la homeostasis de la glucosa intracelular. sis, lo que en última instancia conduce a la acumulación de glucógeno y lípidos mulación en hepatocitos, con clínico manifestaciones que van desde insuficiencia hepática hasta hepatomegalia con alto riesgo de transformación maligna. 125 Por lo tanto, GSD1a se acompaña de alteraciones en la metabolismo del cobre causado por una mutación en ATP7B señalización de AMPK y MTORC1 (Figura 2), así como por la (ATPasa transportadora de cobre beta) que promueve la regulación a la baja de varios componentes centrales de la acumulación de cobre en múltiples órganos, incluidos el maquinaria de la autofagia, lo que apunta a amplios hígado, los riñones y los ojos.122 Atp7b- / - Las células tratadas defectos en los programas de transcripción para la con cobre regulan positivamente un panel de 103 genes regulación de la autofagia.126 De acuerdo con esta noción, relacionados con la autofagia y las funciones lisosomales, y GSD1a se ha asociado con una señalización defectuosa de los estudios de microscopía electrónica confirmaron la sirtuina 1 (SIRT1),127 cuales Revista de Hepatología 2020 vol. 72j 183–196 189 Revisar compromete la actividad de TFEB. Es de destacar que la activación de la autofagia por medios farmacológicos o genéticos limita la acumulación de glucógeno y lípidos en modelos celulares y animales de GSD1a.126 que migran a áreas de lesión tisular y secretan i) Sin embargo, hasta el momento no se han desarrollado beta 1 (TGFB1).141 Por lo tanto, la desaparición continua de paradigmas terapéuticos basados en este enfoque. los hepatocitos provocada por la lesión hepática crónica componentes de ECM, incluido el colágeno de tipo I (en apoyo de la cicatrización de heridas), y ii) citocinas fibrogénicas como el factor de crecimiento transformante fomenta una respuesta de curación de heridas progresiva y Puntos clave Múltiples instancias de autofagia de carga selectiva desempeñan un papel fundamental en la preservación del hígado homeostasis. Enfermedad del hígado graso no alcohólico que no se resuelve y que culmina en una fibrosis extensa.142 La pandemia actual de obesidad y diabetes ha provocado un aumento considerable en la incidencia de NAFLD, con manifestaciones clínicas que van desde la esteatosis simple hasta NASH y un elevado potencial de transformación maligna.49 NAFLD se caracteriza por la acumulación de lípidos dentro del RE de los hepatocitos, 128 que conduce a la muerte celular y daño hepático agudo con un alto potencial de conversión en enfermedad hepática crónica.49 La evidencia acumulada indica que la autofagia (muy probablemente la lipofagia) contrarresta enérgicamente la NAFLD y puede quedar inhabilitada, al menos en parte, durante la patogénesis de la NAFLD. NAFLD se asocia a menudo con la presencia de "megamitocondria",129 lo más probable es que refleje una mitofagia alterada.130 Además, múltiples proteínas ATG y TFEB están reguladas a la baja en los hepatocitos de pacientes con NASH, así como en los hepatocitos de ratones que reciben una HFD o una dieta deficiente en metionina / colina.23,68 Además, la deleción específica de hepatocitos de genes que codifican reguladores de autofagia esenciales, incluidos Atg7, Atg14, o Tfeb, así como la deleción específica de células endoteliales o mieloides de Atg5 exacerbar la sensibilidad de los ratones para desarrollar NAFLD acompañado de una producción elevada de citocinas proinflamatorias en respuesta a un HFD.131-133 Estas observaciones sugieren que la autofagia contrarresta la EHGNA no solo al preservar la homeostasis de los hepatocitos, sino también al limitar el potencial inflamatorio de las células inmunitarias que se infiltran en el hígado. La autofagia puede tener un doble impacto en la De acuerdo con lo anterior, la sobreexpresión de TFEB impulsada por transgenes en los hepatocitos reduce la gravedad de la enfermedad en ratones expuestos a un HFD.68 Además, se ha demostrado que múltiples activadores farmacológicos de la autofagia median los efectos beneficiosos en modelos celulares o animales de NAFLD. Estos incluyen (pero no se limitan a): restricción calórica,134 ejercicio,135 trehalosa136,137 así como ezetimiba, un fármaco hipolipemiante con potencial activador de AMPK que actualmente se está evaluando en ensayos clínicos por su actividad terapéutica contra la EHNA.138,139 Será interesante ver si finalmente se aprobará ezetimiba para esta indicación. Fibrosis La fibrosis hepática implica la acumulación excesiva de componentes de la matriz extracelular (MEC), incluidos diferentes tipos de colágeno.140 Las células estrelladas hepáticas (HSC) son la principal fuente de colágeno en el hígado y de facto subyacen a la fibrogénesis en el contexto de una lesión hepática crónica.141 En este contexto, las HSC se diferencian en forma fibrogénesis hepática (Figura 8). Por un lado, parece que se requiere una autofagia competente para la transdiferenciación de las HSC, al menos en parte debido a su participación en la lipólisis (las HSC pierden LD en el proceso).143.144 De acuerdo con esto, las HSC con ATG2A agotado no se someten a una transdiferenciación espontánea en cultivo celular.144 Además, la eliminación específica de HSC deAtg5 o Atg7 hace que los ratones sean menos susceptibles a la fibrosis hepática inducida por tetracloruro de carbono.145 Aparentemente en desacuerdo con esta noción, el adaptador autofágico SQSTM1 está regulado a la baja en las HSC transdiferenciadas y su ablación acelera (en lugar de desacelerar) la fibrogénesis.146 Sin embargo, dicha actividad no implica respuestas autofágicas, sino que refleja la capacidad de SQSTM1 para favorecer la activación dependiente de la dimerización del receptor de vitamina D y el retinoide X receptor.146 Por otro lado, la autofagia en hepatocitos, células endoteliales sinusoidales hepáticas (LSEC) y macrófagos media citoprotector, anti- de autofagia de autofagia Hígado sano Transdiferenciación de HSC Producción de colágeno Fibrótico hígado Progresión tumoral Diseminación metastásica Resistencia a la terapia Hepatocelular carcinoma Antiinflamatorio Inhibición de la secreción de IL-1B Antioxidante en LSEC Acumulación de la matriz extracelular Inhibición de malignos transformación Degradación de YAP1, NFE2L2 y SQSTM1 Neoplásico enfermedad Fig. 8. Doble impacto de la autofagia sobre la fibrosis hepática y el carcinoma hepatocelular. Por un lado, la autofagia limita la fibrogénesis hepática y la transformación maligna al mediar funciones antioxidantes y antiinflamatorias en macrófagos y LSEC, así como al degradar proteínas potencialmente oncogénicas. Por otro lado, la autofagia apoya la fibrogénesis al favorecer la transdiferenciación de las CMH y promueve la progresión, diseminación y resistencia a la terapia de los carcinomas hepatocelulares establecidos. HSC, célula estrellada hepática; IL1B, interleucina 1 beta; LSEC, célula endotelial sinusoidal hepática; NFE2L2, factor nuclear, eritroide 2 como 2; SQSTM1, secuestosoma 1; YAP1, Sí proteína asociada 1. trans hacia células similares a los miofibroblastos. 190 Papel protector Papel negativo Revista de Hepatología 2020 vol. 72j 183–196 DIARIO DE HEPATOLOGÍA efectos inflamatorios y antioxidantes que limitan la lesión hepática y, por tanto, suprimen el inicio de la fibrosis.131.147– 149 De acuerdo con esta noción, la deleción específica de hepatocitos o macrófagos deAtg5,147,148 así como la eliminación específica de LSEC de Atg7,131 acelera la progresión de la enfermedad en varios modelos de fibrosis en roedores. Es de destacar que en los hepatocitos y las LSEC, la autofagia opera principalmente como un mecanismo antioxidante y citoprotector que preserva la obtenido en roedores inmunodeficientes injertados con células cancerosas humanas y, por lo tanto, no consideran el papel clave de la autofagia en la inmunidad contra el cáncer.165,166 Además, los moduladores farmacológicos de la autofagia actualmente disponibles adolecen de problemas de especificidad.59 Por tanto, se requiere investigación adicional antes de que los moduladores de la autofagia puedan traducirse en agentes clínicos contra el CHC. homeostasis celular.131,147,149 mientras que en los macrófagos y las células de Kupffer, la actividad antifibrótica de la autofagia proviene de la inhibición de la activación del inflamasoma y la consiguiente secreción de la citoquina proinflamatoria y fibrogénica interleucina 1 beta (IL1B).150,151 En conjunto, estas observaciones sugieren que la autofagia tiene un impacto dependiente del contexto sobre la fibrosis hepática, lo que complica el desarrollo de paradigmas terapéuticos basados en la modulación de la autofagia. Hiperamonemia El hígado es un sitio importante para la desintoxicación de productos del catabolismo de proteínas que contienen nitrógeno. Por tanto, la disfunción hepatocelular y Los defectos genéticos en las enzimas del ciclo de la urea pueden conducir a hiperamonemia sistémica y encefalopatía hepática, una condición que confiere un alto riesgo de coma y muerte si no se trata.167 El amoníaco desencadena una rápida respuesta autofágica.168 que se dirige preferentemente a las mitocondrias de una manera que no depende de unc-51 como la autofagia que activa la quinasa Carcinoma hepatocelular 1 (ULK1),169 pero involucra la desacetilasa sirtuina 5 (SIRT5), El CHC es la neoplasia hepática más común y representa 170 así aproximadamente el 90% de todas las neoplasias malignas clave para la autofagia en la depuración de amoníaco, hepáticas.152 En este contexto, la transformación maligna ratones con una deleción de hígado específica de Atg7 o la puede originarse a partir de un panel de diversas pérdida de la función de TFEB presentan una como la inhibición de MTORC1.171 Apoyando un papel alteraciones genéticas que afectan a las cascadas de desintoxicación deficiente del amoníaco.171 Por el contrario, transducción de señales clave, como PI3K? AKT1? MTORC1, la activación de la autofagia en el hígado, inducida por la RAS? RAF? MAPK y WNT?B Vías de catenina.152 sobreexpresión de TFEB impulsada por transgén o la Reflejando su papel clave en la preservación de la administración de rapamicina, limita la acumulación de homeostasis genética,153 Las respuestas autofágicas amoníaco en la circulación de los ratones.171 competentes en los hepatocitos limitan la transformación Por tanto, la activación de la autofagia se destaca como un maligna (Figura 8).22 De acuerdo con esta noción, los enfoque terapéutico prometedor para la hiperamonemia ratones que tienen una deleción en mosaico de Atg5 o una tanto heredada como adquirida. deleción específica de hepatocitos de Atg7 desarrollan espontáneamente múltiples adenomas hepáticos a medida Infecciones virales que envejecen.154 Además, la autofagia parece limitar la El virus de la hepatitis B (VHB) y el virus de la hepatitis C transformación maligna en el hígado al degradar la proteína (VHC) son virus hepatotrópicos de alta prevalencia que 1 asociada a Yes (YAP1),155 un transductor clave de la pueden establecer infecciones crónicas que culminan en señalización de Hippo con un papel importante en la cirrosis y CHC.172,173 Como muchos otros virus, tanto el VHB oncogénesis hepática.156 Aparentemente en desacuerdo con como el VHC activan de forma potente la xenofagia como esto, SQSTM1 parece promover, en lugar de inhibir, la primera línea de defensa hepatocelular.26 La autofagia carcinogénesis hepática.157 El mecanismo subyacente, sin impulsada por el VHB y el VHC se inicia en gran medida por embargo, no está relacionado con la activación de la la respuesta al estrés del RE que resulta de la traducción autofagia,28 y más bien refleja la capacidad de SQSTM1 para incontrolada de proteínas virales.174-176 Además, la proteína favorecer la activación del factor nuclear, eritroide 2 como 2 VHB X (HBx) favorece la autofagia al promover BECN1 (NFE2L2, mejor conocido como NRF2).158–160 transactivación,177 mientras que múltiples proteínas Por el contrario, en los CHC establecidos, la autofagia favorece la progresión tumoral, la diseminación metastásica y la resistencia a la terapia. 22,161 Por tanto, varios inhibidores lisosomales que incluyen cloroquina e hidroxicloroquina median la actividad terapéutica como agentes independientes o combinados con quimioterapia, radioterapia o agentes anticancerígenos dirigidos en una variedad de modelos de CHC de roedores.162-164 Estos hallazgos apuntan a la idea de que la autofagia podría constituir un objetivo potencial para el desarrollo de nuevos regímenes terapéuticos contra el CHC (Figura 8).20 Sin embargo, la mayoría de los datos en apoyo de esta noción se han codificadas en el genoma del VHC, que incluyen p7, NS3 / 4A y NS4B, impulsan la autofagia al interactuar directa o indirectamente con los componentes centrales de la maquinaria de la autofagia, incluyendo BECN1, VPS34, ATG5 y LC3.178–180 Ambos HBV y el VHC, sin embargo, han desarrollado estrategias para aprovechar la autofagia para la replicación y diseminación viral.28 De acuerdo con esta noción, se ha demostrado que la inhibición de la autofagia por intervención farmacológica o genética limita el rendimiento viral en una variedad de entornos experimentales.181-183 En resumen, la autofagia se destaca como un objetivo prometedor para las infecciones por VHB y VHC. Revista de Hepatología 2020 vol. 72j 183–196 191 Revisar Conclusiones y perspectivas El impacto de la autofagia en la fisiopatología hepática acaba de comenzar a surgir, lo que implica que se requieren investigaciones adicionales para traducir datos preclínicos (Nueva York, EE. UU.), Y mediante donaciones de Phosplatin (Nueva York, EE. UU.), La Fundación Luke Heller TECPR2 (Boston, EE. UU.) Y Sotio as (Praga, República Checa). CH está financiado por FONDECYT no. y prometedores sobre la modulación de la autofagia en 1140549 estrategias terapéuticas que se puedan utilizar en la clínica. 15150012, Millennium Institute P09-015-F, Comisión 20 Para Europea I + D MSCA-RISE # 734749, y agradecemos el apoyo ello, será de suma importancia analizar la 1180186, FONDAP programa complejidad de la red autofágica en modelos de de Michael J Fox Foundation for Parkinson's Research - enfermedades específicas, con especial énfasis en los Target Validation grant No 9277, FONDEF ID16I10223, efectos potencialmente antagonistas de las respuestas FONDEF D11E1007, Oficina de Investigación Naval de EE. autofágicas en los diferentes tipos de células del UU. - Global (ONR-G) N62909-16-1-2003, Oficina de microecosistema hepático. El desarrollo de moduladores de Investigación Científica de la Fuerza Aérea de EE. UU. autofagia altamente específicos y plataformas moleculares FA9550-16-1-0384, ALSRP Thera- para la administración dirigida también se destaca como un peutico desafío importante para la traducción clínica de este Asociación de Distrofia 382453 y CONICYT Brasil 441921 / 2016-7. GK cuenta con el apoyo de la Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) - Projets blancs; ANR en el marco de E-Rare-2, ERA-Net para la investigación de enfermedades raras; Association pour la recherche sur le cancer (ARC); Cancéropôle Ilede-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); una donación de Elior; Red del Espacio Europeo de Investigación en Enfermedades Cardiovasculares (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, Oncobiome del Proyecto Horizonte 2020 de la Unión Europea; Fundación Carrefour; Programa de expertos extranjeros de alto nivel en China (GDW20171100085 y paradigma.20 A pesar de estos y otros obstáculos, el potencial terapéutico de los moduladores de la autofagia para el tratamiento de múltiples trastornos hepáticos sigue siendo alto, lo que refleja en gran medida el papel central de las respuestas autofágicas coordinadas en la preservación de la homeostasis hepática. Nuestra esperanza es que la carbamazepina y la ezetimiba se conviertan en los primeros de una larga lista de moduladores de la autofagia que se utilizarán para el tratamiento de pacientes con enfermedad hepática, inaugurando así una historia de éxito clínico. Abreviatura 3-MA, 3-metiladenina; CMA, acompañante autofagia mediada; ECM, matriz extracelular; RE, retículo endoplásmico; ERAD, degradación asociada al retículo endoplásmico; FA, ácido graso; FAO, oxidación de ácidos grasos; GSD1a, enfermedad por almacenamiento de glucógeno tipo 1a; HCC, carcinoma hepatocelular; HFD, dieta rica en grasas; HSC, célula estrellada hepática; IMM, membrana mitocondrial interna; LD, gota de lípido; LIR, región que interactúa con LC3; LSEC, célula endotelial Idea Otorgar AL150111, Muscular GDW20181100051), Institut National du Cancer (Inca); Inserm (HTE); Inserm Transfert, Institut Universitaire de France; Fundación LeDucq; los LabEx Immuno-Oncology; el RHU Torino Lumière; la Fundación Seerave; SIRIC Reparación de ADN de células oncológicas estratificadas y eliminación inmunitaria de tumores (SOCRATE); y el SIRIC Investigación del cáncer y medicina personalizada (CARPEM). sinusoide hepática; NAFLD, enfermedad del hígado graso no alcohólico; NASH, esteatohepatitis no alcohólica; OMM, membrana mitocondrial externa; PKA, proteína quinasa A; Conflicto de intereses RHD, dominio de homología de retículo; ROS, especies Los autores declaran no tener ningún conflicto de intereses reactivas de oxígeno; SAM,S- relacionado con este trabajo. adenosil-L-metionina; SOCE, Ca operado por la tienda2+ entrada; TCPOBOP, 1,4-bis [2- (3,5dicloropiridiloxi)] benceno. Consulte el adjunto Divulgación del ICMJE formularios para obtener más detalles. Soporte financiero YH está financiado por FONDECYT no. 3180427. LG cuenta con el Dato suplementario Se pueden encontrar datos complementarios a este artículo en línea en https://doi.org/10.1016/j.jhep.2019.08. 026. apoyo de una subvención Breakthrough Level 2 del Departamento de Defensa de EE. UU., Programa de Investigación del Cáncer de Mama (BCRP) [# BC180476P1], de una subvención inicial del Departamento de Oncología Radioterápica de Weill Cornell Medicine (Nueva York, EE. UU.), por colaboraciones industriales con Lytix (Oslo, Noruega) y Phosplatin 192 Revista de Hepatología 2020 vol. 72j 183–196 DIARIO DE HEPATOLOGÍA Referencias [1] Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Definiciones moleculares de autofagia y procesos relacionados. EMBO J 2017; 36: 1811–1836. [2] Dikic I, Elazar Z. Mecanismo e implicaciones médicas de la autofagia de mamíferos. Nat Rev Mol Cell Biol 2018; 19: 349–364. [3] De Duve C, Wattiaux R. Funciones de los lisosomas. Annu Rev Physiol 1966; 28: 435–492. [4] Appelmans F, Wattiaux R, De Duve C. Estudios de fraccionamiento de tejidos. 5. La asociación de la fosfatasa ácida con una clase especial de gránulos citoplasmáticos en el hígado de rata. Biochem J 1955; 59: 438–445. [5] De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F.Estudios de fraccionamiento de tejidos. 6. Patrones de distribución intracelular de enzimas en tejido de hígado de rata. Biochem J 1955; 60: 604–617. [6] Novikoff AB, Beaufay H, De Duve C. Microscopía electrónica de fracciones lisosoméricas de hígado de rata. J Biophys Biochem Cytol 1956; 2: 179–184. [7] Beaufay H, Bendall DS, Baudhun P, Wattiaux R, De Duve C. Estudios de fraccionamiento de tejidos. 13. Análisis de fracciones mitocondriales de hígado de rata mediante centrifugación en gradiente de densidad. Biochem J 1959; 73: 628–637. [8] Straus W. Identificación citoquímica rápida de fagosomas en varios tejidos de la rata y su diferenciación de las mitocondrias por el método de la peroxidasa. J Biophys Biochem Cytol 1959; 5: 193–204. [9] Essner E, Novikoff AB. Localización de la actividad de la fosfatasa ácida en lisosomas hepáticos mediante microscopía electrónica. J Biophys Biochem Cytol 1961; 9: 773–784. [10] Ashford TP, Porter KR. Componentes citoplásmicos en lisosomas de células hepáticas. J Cell Biol 1962; 12: 198–202. [11] Moe H, Behnke O. Cuerpos citoplasmáticos que contienen mitocondrias, ribosomas y membranas endoplásmicas de superficie rugosa en el epitelio del intestino delgado de ratas recién nacidas. J Cell Biol 1962; 13: 168-171. [12] Straus W. Observaciones citoquímicas sobre la relación entre lisosomas y fagosomas en riñón e hígado mediante tinción combinada para fosfatasa ácida y peroxidasa de rábano picante inyectada por vía intravenosa. J Cell Biol 1964; 20: 497–507. [13] Miller LL. Glucagón: una hormona catabólica proteica en el hígado de rata perfundido aislado. Naturaleza 1960; 185: 248. [14] Kaushik S, Cuervo AM. La mayoría de edad de la autofagia mediada por acompañantes. Nat Rev Mol Cell Biol 2018; 19: 365–381. [15] Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, Kroemer G. Organelle-specific iniciation of autphagy. Mol Cell 2015; 59: 522–539. [dieciséis] Farre JC, Subramani S. Conocimientos mecanicistas sobre las vías de autofagia selectiva: lecciones de la levadura. Nat Rev Mol Cell Biol 2016; 17: 537–552. [17] Levine B, Kroemer G. Funciones biológicas de los genes de la autofagia: una perspectiva de la enfermedad. Cell 2019; 176: 11–42. [18] Galluzzi L, Yamazaki T, Kroemer G. Vincular las respuestas de estrés celular a la homeostasis sistémica. Nat Rev Mol Cell Biol 2018; 19: 731–745. [19] Ueno T, Komatsu M. Autofagia en el hígado: funciones en la salud y la enfermedad. Nat Rev Gastroenterol Hepatol 2017; 14: 170–184. [20] Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autofagia en enfermedades hepáticas: ¿tiempo de traducción ?. J Hepatol 2019; 70: 985–998. [21] Kroemer G, Marino G, Levine B. La autofagia y la respuesta integrada al estrés. Mol Cell 2010; 40: 280-293. [22] Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autofagia en la transformación maligna y progresión del cáncer. EMBO J 2015; 34: 856–880. [23] Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. La autofagia regula el metabolismo de los lípidos. Nature 2009; 458: 1131–1135. [24] Galluzzi L, Pietrocola F, Levine B, Kroemer G. Control metabólico de la autofagia. Cell 2014; 159: 1263–1276. [25] Mizushima N, Levine B, Cuervo AM, Klionsky DJ. La autofagia combate las enfermedades a través de la autodigestión celular. Nature 2008; 451: 1069–1075. [26] Deretic V, Levine B. La autofagia equilibra la inflamación en la inmunidad innata. Autofagia 2018; 14: 243–251. [27] Xie M, Yang Z, Liu Y, Zheng M. El papel de la autofagia inducida por el VHB en la replicación del VHB y el HCC relacionado con el VHB. Life Sci 2018; 205: 107–112. [28] Galluzzi L, DR Verde. Funciones independientes de la autofagia de la maquinaria de autofagia. Cell 2019; 177: 1682–1699. [29] Ueno T, Ezaki J, Kominami E. Contribución metabólica de la proteólisis autofágica hepática: vino viejo en botellas nuevas. Biochim Biophys Acta 2012; 1824: 51–58. [30] Kirschke H, Langner J, Wiederanders B, Ansorge S, Bohley P. Cathepsin L. Una nueva proteinasa de lisosomas de hígado de rata. Eur J Biochem 1977; 74: 293–301. [31] Schworer CM, Shiffer KA, Mortimore GE. Relación cuantitativa entre la autofagia y la proteólisis durante la privación gradual de aminoácidos en hígado de rata perfundido. J Biol Chem 1981; 256: 7652–7658. [32] Mortimore GE, Hutson Nueva Jersey, Surmacz CA. Correlación cuantitativa entre proteólisis y macro y microautofagia en hepatocitos de ratón durante la inanición y la realimentación. Proc Natl Acad Sci USA 1983; 80: 2179–2183. [33] Anding AL, Baehrecke EH. Limpieza de casa: autofagia selectiva de orgánulos. Dev Cell 2017; 41: 10–22. [34] Hetz C, Papa FR. La respuesta de la proteína desplegada y el control del destino celular. Mol Cell 2018; 69: 169–181. [35] Hetz C, Chevet E, Harding HP. Dirigirse a la respuesta de la proteína desplegada en la enfermedad. Nat Rev Drug Discov 2013; 12: 703–719. [36] Ellgaard L, Sevier CS, Bulleid NJ. ¿Cómo se reducen las proteínas en el retículo endoplásmico ?. Trends Biochem Sci 2018; 43: 32–43. [37] Schwarz DS, soplador MD. El retículo endoplásmico: estructura, función y respuesta a la señalización celular. Cell Mol Life Sci 2016; 73: 79–94. [38] Grumati P, Dikic I, Stolz A. ER-phagy de un vistazo. J Cell Sci 2018; 131: jcs217364. [39] Gatica D, Lahiri V, Klionsky DJ. Reconocimiento y degradación de la carga por autofagia selectiva. Nat Cell Biol 2018; 20: 233–242. [40] Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, et al. RTN3 de longitud completa regula el recambio del retículo endoplásmico tubular mediante autofagia selectiva. Elife 2017; 6. [41] Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulación del recambio del retículo endoplásmico por autofagia selectiva. Nature 2015; 522: 354–358. [42] Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, et al. CCPG1 es un receptor de carga de autofagia no canónico esencial para la proteostasis de ER-fagia y ER pancreática. Dev Cell 2018; 44, 217–32 e11. [43] Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, et al. La autofagia selectiva mediada por receptores degrada el retículo endoplásmico y el núcleo. Nature 2015; 522: 359–362. [44] Yamamoto Y, Sakisaka T. Los factores de biogénesis del peroxisoma apuntan postraduccionalmente a las proteínas que contienen el dominio de homología del retículo a la membrana del retículo endoplásmico. Sci Rep 2018; 8: 2322. [45] Niso-Santano M, Malik SA, Pietrocola F, Bravo-San Pedro JM, Marino G, Cianfanelli V, et al. Los ácidos grasos insaturados inducen la autofagia no canónica. EMBO J 2015; 34: 1025–1041. [46] Pang L, Liu K, Liu D, Lv F, Zang Y, Xie F, et al. Efectos diferenciales de la reticulofagia y la mitofagia sobre la enfermedad del hígado graso no alcohólico. Muerte celular Dis 2018; 9: 90. [47] Yang H, Ni HM, Guo F, Ding Y, Shi YH, Lahiri P, et al. La proteína Sequestosome 1 / p62 se asocia con la eliminación autofágica del exceso de retículo endoplásmico hepático en ratones. J Biol Chem 2016; 291: 18663–18674. [48] Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, et al. Una ER-fagia selectiva ejerce un control de calidad del procolágeno a través de un complejo Calnexin-FAM134B. EMBO J 2019; 38. [49] Gluchowski NL, Becuwe M, Walther TC, Farese Jr RV. Gotas de lípidos y enfermedad hepática: de la biología básica a las implicaciones clínicas. Nat Rev Gastroenterol Hepatol 2017; 14: 343–355. [50] Walther TC, Chung J, Farese Jr RV. Biogénesis de gotitas de lípidos. Annu Rev Cell Dev Biol 2017; 33: 491–510. [51] Prinz WA. Un puente para comprender el crecimiento de las gotas de lípidos. Dev Cell 2013; 24: 335–336. [52] Renvoise B, Malone B, Falgairolle M, Munasinghe J, Stadler J, Sibilla C, et al. Los ratones nulos de Reep1 revelan un papel convergente de las proteínas de la paraplejía espástica hereditaria en la regulación de las gotas de lípidos. Hum Mol Genet 2016; 25: 5111–5125. [53] Wilfling F, Thiam AR, Olarte MJ, Wang J, Beck R, Gould TJ, et al. La maquinaria Arf1 / COPI actúa directamente sobre las gotas de lípidos y permite su conexión con el RE para la selección de proteínas. Elife 2014; 3 e01607. [54] Soni KG, Mardones GA, Sougrat R, Smirnova E, Jackson CL, Bonifacino JS. Entrega de proteínas dependientes de coatómeros a gotitas de lípidos. J Cell Sci 2009; 122: 1834–1841. [55] Houten SM, Violante S, Ventura FV, Wanders RJ. La bioquímica y fisiología de la beta-oxidación de ácidos grasos mitocondriales y sus trastornos genéticos. Annu Rev Physiol 2016; 78: 23–44. [56] Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetil coenzima A: un metabolito central y segundo mensajero. Cell Metab 2015; 21: 805–821. [57] Zechner R, Madeo F, Kratky D. Lipólisis citosólica y lipofagia: dos caras de la misma moneda. Nat Rev Mol Cell Biol 2017; 18: 671–684. [58] Rui L. Metabolismo energético en el hígado. Compr Physiol 2014; 4: 177–197. Revista de Hepatología 2020 vol. 72j 183–196 193 Revisar [59] Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Modulación farmacológica de la autofagia: potencial terapéutico y obstáculos persistentes. Nat Rev Drug Discov 2017; 16: 487–511. [81] Lee J, Homma T, Kobayashi S, Ishii N, Fujii J. Revelación de los trastornos orgánicos sistémicos asociados con el catabolismo de lípidos alterado en ratones deficientes en SOD1 en ayunas. Arch Biochem Biophys 2018; 654: 163-171. [60] Schulze RJ, Drizyte K, Casey CA, McNiven MA. La lipofagia hepática: nuevos conocimientos sobre el catabolismo autofágico de las gotitas de lípidos en el hígado. Hepatol Commun 2017; 1: 359–369. [82] Youle RJ, Narendra DP. Mecanismos de la mitofagia. Nat Rev Mol Cell Biol 2011; 12: 9–14. [61] Zubiete-Franco I, García-Rodríguez JL, Martínez-Una M, Martínez-López N, Woodhoo A, Juan VG, et al. Los niveles de metionina y S-adenosilmetionina son reguladores críticos de la actividad de PP2A que modula la lipofagia durante la esteatosis. J Hepatol 2016; 64: 409–418. [62] Lettieri Barbato D, Tatulli G, Aquilano K, Ciriolo MR. FoxO1 controla la lipasa ácida lisosómica en los adipocitos: implicación de la lipofagia durante la restricción de nutrientes y el tratamiento con metformina. Muerte celular Dis 2013; 4 e861. [63] Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, et al. Dirigirse al factor de transcripción UPR XBP1 protege contra la enfermedad de Huntington mediante la regulación de FoxO1 y la autofagia. Hum Mol Genet 2012; 21: 2245–2262. [64] Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. La pequeña GTPasa Rab7 como regulador central de la lipofagia hepatocelular. Hepatología 2015; 61: 1896-1907. [sesenta y cinco] Schulze RJ, Rasineni K, Weller SG, Schott MB, Schroeder B, Casey CA, et al. La exposición al etanol inhibe la lipofagia de los hepatocitos al inactivar la pequeña guanosina trifosfatasa Rab7. Hepatol Commun 2017; 1: 140–152. [66] Li C, Luo X, Zhao S, Siu GK, Liang Y, Chan HC, et al. COPI-TRAPPII activa Rab18 y regula su asociación de gotitas de lípidos. EMBO J 2017; 36: 441– 457. [67] Li Z, Schulze RJ, Weller SG, Krueger EW, Schott MB, Zhang X, et al. Un novedoso complejo Rab10-EHBP1-EHD2 esencial para la absorción autofágica de gotitas de lípidos. Sci Adv 2016; 2 e1601470. [68] Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controla el metabolismo de los lípidos celulares a través de un bucle autorregulador inducido por la inanición. Nat Cell Biol 2013; 15: 647–658. [69] Kaushik S, Cuervo AM. La degradación de proteínas asociadas a gotitas de lípidos por autofagia mediada por chaperonas facilita la lipólisis. Nat Cell Biol 2015; 17: 759–770. [70] Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, et al. La entrada de Ca (2+) operada por el almacén controla la inducción de la lipólisis y la reprogramación transcripcional del metabolismo de los lípidos. Cell Metab 2017; 25: 698–712. [71] Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, et al. La cafeína estimula el metabolismo de los lípidos hepáticos mediante la vía autofagialisosomal en ratones. Hepatología 2014; 59: 1366–1380. [72] Ding WX. Beber café quema la grasa hepática al inducir lipofagia junto con la beta-oxidación mitocondrial. Hepatología 2014; 59: 1235–1238. [73] Pietrocola F, Malik SA, Marino G, Vacchelli E, Senovilla L, Chaba K, et al. El café induce la autofagia in vivo. Ciclo celular 2014; 13: 1987–1994. [74] Brandt A, Nier A, Jin CJ, Baumann A, Jung F, Ribas V, et al. El consumo de café descafeinado protege contra el desarrollo de esteatohepatitis no alcohólica temprana: papel de la función de barrera intestinal. Redox Biol 2019; 21 101092. [75] Setiawan VW, Wilkens LR, Lu SC, Hernandez BY, Le Marchand L, Henderson BE. Asociación de la ingesta de café con una incidencia reducida de cáncer de hígado y muerte por enfermedad hepática crónica en la cohorte multiétnica de EE. UU. Gastroenterología 2015; 148, 118–25; cuestionario e15. [76] Xiao Q, Sinha R, Graubard BI, Freedman ND. Asociaciones inversas de café total y descafeinado con niveles de enzimas hepáticas en la Encuesta Nacional de Examen de Salud y Nutrición 1999-2010. Hepatología 2014; 60: 2091-2098. [77] Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Mecanismos moleculares de la muerte celular: recomendaciones del Comité de Nomenclatura sobre Muerte Celular 2018. Cell Death Differ 2018; 25: 486–541. [78] Hamlin AN, Basford JE, Jaeschke A, Hui DY. La deficiencia de la proteína LRP1 exacerba la esteatosis inducida por palmitato y la toxicidad en los hepatocitos. J Biol Chem 2016; 291: 16610–16619. [79] Hamlin AN, Chinnarasu S, Ding Y, Xian X, Herz J, Jaeschke A, et al. La disfunción de la proteína 1 relacionada con el receptor de lipoproteínas de baja densidad se sinergiza con el colesterol de la dieta para acelerar la progresión de la esteatohepatitis. J Biol Chem 2018; 293: 9674–9684. [80] Kurahashi T, Hamashima S, Shirato T, Lee J, Homma T, Kang ES, et al. Una deficiencia de SOD1 aumenta la acumulación de gotitas de lípidos en el hígado del ratón en ayunas al interrumpir la lipofagia. Biochem Biophys Res Commun 2015; 467: 866–871. 194 [83] Tatsuta T, Langer T. Control de calidad de las mitocondrias: protección contra la neurodegeneración y el envejecimiento. EMBO J 2008; 27: 306–314. [84] Saito T, Sadoshima J. Mecanismos moleculares de la autofagia / mitofagia mitocondrial en el corazón. Circ Res 2015; 116: 1477–1490. [85] Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. La disfunción mitocondrial en mutantes de Drosophila PINK1 se complementa con parkina. Nature 2006; 441: 1157–1161. [86] Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 es necesaria para la función mitocondrial e interactúa genéticamente con la parkina. Nature 2006; 441: 1162–1166. [87] Lazarou M, Jin SM, Kane LA, Youle RJ. Papel de la unión de PINK1 al complejo TOM y membranas intracelulares alternas en el reclutamiento y activación de la ligasa E3 Parkin. Dev Cell 2012; 22: 320–333. [88] Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, et al. La autofosforilación de PINK1 tras la disipación del potencial de membrana es esencial para el reclutamiento de Parkin en las mitocondrias dañadas. Nat Commun 2012; 3: 1016 . [89] Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. La ubiquitina es fosforilada por PINK1 para activar la parkina. Nature 2014; 510: 162–166. [90] Chen Y, Dorn 2nd GW. La mitofusina 2 fosforilada PINK1 es un receptor de Parkin para eliminar las mitocondrias dañadas. Science 2013; 340: 471–475. [91] Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autofagia y mitofagia en la enfermedad cardiovascular. Circ Res 2017; 120: 1812–1824. [92] Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet-Rousselle C, et al. La externalización de cardiolipina mediada por NDPK-D (NM23-H4) permite la eliminación de las mitocondrias despolarizadas por mitofagia. Diferencia de muerte celular 2016; 23: 1140–1151. [93] Kim I, Lemasters JJ. Degradación mitocondrial por autofagia (mitofagia) en hepatocitos transgénicos GFP-LC3 durante la privación de nutrientes. Am J Physiol Cell Physiol 2011; 300: C308 – C317. [94] Lemasters JJ. Autofagia mitocondrial selectiva, o mitofagia, como defensa dirigida contra el estrés oxidativo, la disfunción mitocondrial y el envejecimiento. Rejuvenation Res 2005; 8: 3–5. [95] Kim I, Lemasters JJ. La mitofagia degrada selectivamente las mitocondrias dañadas individuales después de la fotoirradiación. Señal redox antioxidante 2011; 14: 1919-1928. [96] Kim I, Rodríguez-Enriquez S, Lemasters JJ. Degradación selectiva de mitocondrias por mitofagia. Arch Biochem Biophys 2007; 462: 245–253. [97] Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin se recluta selectivamente para las mitocondrias deterioradas y promueve su autofagia. J Cell Biol 2008; 183: 795–803. [98] Kroemer G, Galluzzi L, Brenner C.Permeabilización de la membrana mitocondrial en la muerte celular. Physiol Rev 2007; 87: 99–163. [99] Poole B. Biogénesis y recambio de peroxisomas de hígado de rata. Ann NY Acad Sci 1969; 168: 229–243. [100] Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, et al. ATM funciona en el peroxisoma para inducir pexofagia en respuesta a ROS. Nat Cell Biol 2015; 17: 1259–1269. [101] Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, et al. NBR1 actúa como receptor de autofagia para peroxisomas. J Cell Sci 2013; 126: 939– 952. [102] Sargent G, van Zutphen T, Shatseva T, Zhang L, Di Giovanni V, Bandsma R, et al. PEX2 es la ubiquitina ligasa E3 necesaria para la pexofagia durante la inanición. J Cell Biol 2016; 214: 677–690. [103] Eun SY, Lee JN, Nam IK, Liu ZQ, So HS, Choe SK, et al. PEX5 regula la autofagia a través del eje mTORC1-TFEB durante la inanición. Exp Mol Med 2018; 50: 4. [104] Walter KM, Schonenberger MJ, Trotzmuller M, Horn M, Elsasser HP, Moser AB, et al. Hif-2alpha promueve la degradación de los peroxisomas de mamíferos por autofagia selectiva. Cell Metab 2014; 20: 882–897. [105] Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, et al. El exceso de peroxisomas se degrada por la maquinaria autofágica en los mamíferos. J Biol Chem 2006; 281: 4035–4041. [106] Zientara-Rytter K, Subramani S. Degradación autofágica de peroxisomas en mamíferos. Biochem Soc Trans 2016; 44: 431–440. [107] Godfrey R, Quinlivan R. Trastornos del músculo esquelético de la glucogenólisis y la glucólisis. Nat Rev Neurol 2016; 12: 393–402. Revista de Hepatología 2020 vol. 72j 183–196 DIARIO DE HEPATOLOGÍA [108] Zhao H, Tang M, Liu M, Chen L. Glicofagia: un objetivo emergente en patología. Clin Chim Acta 2018; 484: 298–303. desarrollo de esteatohepatitis no alcohólica y fibrosis. J Hepatol 2018; 68: S29-S. [109] Jiang S, Wells CD, Roach PJ. Proteína 1 que contiene el dominio de unión al almidón (Stbd1) y metabolismo del glucógeno: identificación del motivo de interacción de la familia Atg8 (AIM) en Stbd1 necesario para la interacción con GABARAPL1. Biochem Biophys Res Commun 2011; 413: 420–425. [132] Xiong X, Tao R, DePinho RA, Dong XC. El gen 14 relacionado con la autofagia (Atg14) está [110] Devos P, el suyo HG. Glucogenólisis aleatoria, presumiblemente hidrolítica y lisosomal en el hígado de ratas tratadas con florizina y de ratas recién nacidas. Biochem J 1980; 192: 177–181.. [111] David H, Ellermann J, Bimmler M, Behrisch D. Ultraestructura del hígado después de la hipoxia en el período posnatal. Exp Pathol 1991; 43: 97–110. [133] Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. La autofagia de macrófagos alterada aumenta la respuesta inmune en ratones obesos al promover la polarización de macrófagos proinflamatorios. Autofagia 2015; 11: 271–284. [134] Kim KE, Jung Y, Min S, Nam M, Heo RW, Jeon BT, et al. La restricción calórica de ratones db / db revierte la esteatosis hepática y el peso corporal con metabolismo hepático divergente. Sci Rep 2016; 6: 30111. [112] Sun T, Yi H, Yang C, Kishnani PS, Sun B. El dominio de unión al almidón que contiene la proteína 1 juega un papel dominante en el transporte de glucógeno a los lisosomas en el hígado. J Biol Chem 2016; 291: 16479–16484. [113] Hazari YM, Bashir A, Habib M, Bashir S, Habib H, Qasim MA, et al. Deficiencia de alfa-1-antitripsina: variaciones genéticas, manifestaciones clínicas e intervenciones terapéuticas. Mutat Res 2017; 773: 14–25. [114] Lindblad D, Blomenkamp K, Teckman J. El contenido de proteína Z mutante de alfa-1antitripsina en hepatocitos individuales se correlaciona con la muerte celular en un modelo de ratón. Hepatología 2007; 46: 1228–1235. regulado por factores de transcripción de la caja O en forma de horquilla y los ritmos circadianos y desempeña un papel fundamental en la autofagia hepática y el metabolismo de los lípidos. J Biol Chem 2012; 287: 39107–39114. [135] Goncalves IO, Passos E, Diogo CV, Rocha-Rodrigues S, Santos-Alves E, Oliveira PJ, et al. El ejercicio mitiga las alteraciones de los mecanismos de control de calidad y de los poros de transición de la permeabilidad mitocondrial en la esteatohepatitis no alcohólica. Appl Physiol Nutr Metab 2016; 41: 298–306. [136] DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, et al. La trehalosa inhibe las proteínas portadoras de soluto 2A (SLC2A) para inducir la autofagia y prevenir la esteatosis hepática. Sci Signal 2016; 9: ra21. [115] Kroeger H, Miranda E, MacLeod I, Perez J, Crowther DC, Marciniak SJ, et al. La degradación asociada al retículo endoplásmico (ERAD) y la autofagia cooperan para degradar las serpinas mutantes polimerogénicas. J Biol Chem 2009; 284: 22793–22802. [116] Stoller JK, Aboussouan LS. Deficiencia de alfa1-antitripsina. Lancet 2005; 365: 2225–2236. [117] Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, et al. Las inclusiones intracelulares que contienen alfa1-antitripsina Z mutante se propagan en ausencia de actividad autofágica. J Biol Chem 2006; 281: 4467–4476. [118] Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Autofagia mitocondrial y lesión en el hígado en la deficiencia de alfa 1-antitripsina. Am J Physiol Gastrointest Liver Physiol 2004; 286: G851 – G862. [137] Mardones P, Rubinsztein DC, Hetz C. Misterio resuelto: la trehalosa inicia la autofagia al bloquear el transporte de glucosa. Sci Signal 2016; 9: fs2. [119] Teckman JH, Perlmutter DH. La retención de alfa (1) -antitripsina Z mutante en el retículo endoplásmico se asocia con una respuesta autofágica. Am J Physiol Gastrointest Liver Physiol 2000; 279: G961 – G974. [141] Tsuchida T, Friedman SL. Mecanismos de activación de las células estrelladas hepáticas. Nat Rev Gastroenterol Hepatol 2017; 14: 397–411. [142] Mallat A, Lotersztajn S. Mecanismos celulares de la fibrosis tisular. 5. Nuevos conocimientos sobre la fibrosis hepática. Am J Physiol Cell Physiol 2013; 305: C789 – C799. [143] Thoen LF, Guimaraes EL, Dolle L, Mannaerts I, Najimi M, Sokal E, et al. Un papel de la autofagia durante la activación de las células estrelladas hepáticas. J Hepatol 2011; 55: 1353–1360. [138] Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, et al. Ezetimiba mejora la esteatohepatitis a través de la activación de la autofagia mediada por la proteína quinasa activada AMP-TFEB y la inhibición del inflamasoma NLRP3. Autofagia 2017; 13: 1767–1781. [139] Lee DH, Han DH, Nam KT, Park JS, Kim SH, Lee M, et al. Ezetimiba, un inhibidor de NPC1L1, es un potente activador de Nrf2 que protege a los ratones de la esteatohepatitis no alcohólica inducida por la dieta. Free Radical Biol Med 2016; 99: 520–532. [140] Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Reparación y fibrosis hepática: regulación inmunitaria de la cicatrización de heridas en un órgano sólido. Nat Rev Immunol 2014; 14: 181–194. [120] Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, et al. La transferencia de genes del regulador maestro de la autofagia TFEB da como resultado la eliminación de proteínas tóxicas y la corrección de la enfermedad hepática en la deficiencia de alfa-1-anti-tripsina. EMBO Mol Med 2013; 5: 397–412. [121] Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. Un fármaco potenciador de la autofagia promueve la degradación de la alfa1-antitripsina Z mutante y reduce la fibrosis hepática. Science 2010; 329: 229–232. [122] Czlonkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, et al. Enfermedad de Wilson. Nat Rev Dis Primers 2018; 4:21. [123] Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, et al. La activación de la autofagia, observada en tejidos hepáticos de pacientes con enfermedad de Wilson y de animales deficientes en ATP7B, protege a los hepatocitos de la apoptosis inducida por cobre. Gastroenterología 2019; 156, 1173–89 e5. [124] Zischka H, Lichtmannegger J, Schmitt S, Jagemann N, Schulz S, Wartini D, et al. Reticulación y destrucción de la membrana mitocondrial del hígado en un modelo de rata de la enfermedad de Wilson. J Clin Invest 2011; 121: 1508–1518. [125] Farah BL, Sinha RA, Wu Y, Singh BK, Lim A, Hirayama M, et al. La disfunción mitocondrial hepática es una característica de la enfermedad por almacenamiento de glucógeno tipo Ia (GSDIa). Sci Rep 2017; 7: 44408. [126] Farah BL, Landau DJ, Sinha RA, Brooks ED, Wu Y, Fung SYS, et al. La inducción de la autofagia mejora el metabolismo de los lípidos hepáticos en la deficiencia de glucosa-6-fosfatasa. J Hepatol 2016; 64: 370–379. [127] Cho JH, Kim GY, Pan CJ, Anduaga J, Choi EJ, Mansfield BC, et al. La regulación a la baja de la señalización de SIRT1 es la base del deterioro de la autofagia hepática en la enfermedad por almacenamiento de glucógeno tipo Ia. PLoS Genet 2017; 13 e1006819. [128] Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Señalización del estrés del retículo endoplásmico y patogenia de la enfermedad del hígado graso no alcohólico. J Hepatol 2018; 69: 927–947. [129] Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, et al. Anomalías mitocondriales en la esteatohepatitis no alcohólica. J Hepatol 1999; 31: 430–434. [130] Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, et al. La estasis mitocondrial revela ubiquitinación mediada por p62 en la mitofagia independiente de Parkin y mitiga la enfermedad del hígado graso no alcohólico. Cell Metab 2018; 28, 588-604 e5. [131] Hammoutene A, Lasselin J, Vion AC, Colnot N, Paradis V, Lotersztajn S, et al. La autofagia defectuosa en las células endoteliales sinusoidales del hígado promueve [144] Hong Y, Li S, Wang J, Li Y. In vitro inhibición de la activación de células estrelladas hepáticas por la proteína de gota de lípidos relacionada con la autofagia ATG2A. Ciencia Rep 2018; 8: 9232. [145] Hernandez-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, et al. El estrés del retículo endoplásmico induce actividad fibrogénica en las células estrelladas hepáticas a través de la autofagia. J Hepatol 2013; 59: 98–104. [146] Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, et al. p62 / SQSTM1 al unirse al receptor de vitamina D inhibe la actividad de las células estrelladas hepáticas, la fibrosis y el cáncer de hígado. Cáncer Cell 2016; 30: 595–609. [147] Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP y col. Nrf2 promueve el desarrollo de fibrosis y tumorigénesis en ratones con autofagia hepática defectuosa. J Hepatol 2014; 61: 617–625. [148] Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, et al. La autofagia de macrófagos protege contra la fibrosis hepática en ratones. Autofagia 2015; 11: 1280–1292. [149] Ruart M, Chavarria L, Camprecios G, Suarez-Herrera N, Montironi C, GuixeMuntet S, et al. La autofagia endotelial alterada promueve la fibrosis hepática al agravar la respuesta al estrés oxidativo durante la lesión hepática aguda. J Hepatol 2019; 70: 458–469. [150] Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. El bloqueo del inflamasoma NLRP3 reduce la inflamación y la fibrosis del hígado en la EHNA experimental en ratones. J Hepatol 2017; 66: 1037– 1046. [151] Ilyas G, Zhao E, Liu K, Lin Y, Tesfa L, Tanaka KE, et al. La autofagia de macrófagos limita la lesión hepática tóxica aguda en ratones a través de la regulación a la baja de la interleucina-1beta. J Hepatol 2016; 64: 118–127. [152] Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M y col. Carcinoma hepatocelular. Nat Rev Dis Primers 2016; 2: 16018. [153] Rybstein MD, Bravo-San Pedro JM, Kroemer G, Galluzzi L. La red autofágica y el cáncer. Nat Cell Biol 2018; 20: 243–251. [154] Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Los ratones con deficiencia de autofagia desarrollan múltiples tumores hepáticos. Genes Dev 2011; 25: 795–800. Revista de Hepatología 2020 vol. 72j 183–196 195 Revisar [155] Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML, et al. La autofagia es un guardián de la diferenciación hepática y la carcinogénesis al controlar la degradación de Yap. Nat Commun 2018; 9: 4962. [156] Perra A, Kowalik MA, Ghiso E, Ledda-Columbano GM, Di Tommaso L, Angioni MM, et al. La activación de YAP es un evento temprano y un objetivo terapéutico potencial en el desarrollo del cáncer de hígado. J Hepatol 2014; 61: 1088–1096. [170] Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, et al. Regulación SIRT5 de la autofagia y mitofagia inducidas por amoniaco. Autofagia 2015; 11: 253–270. [171] Soria LR, Allegri G, Melck D, Pastore N, Annunziata P, Paris D, et al. La potenciación de la autofagia hepática aumenta la ureagénesis y protege contra la hiperamonemia. Proc Natl Acad Sci USA 2018; 115: 391–396. [157] Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, et al. p62, regulado positivamente durante la preneoplasia, induce carcinogénesis hepatocelular al mantener la supervivencia de las células iniciadoras de CHC estresadas. Cancer Cell 2016; 29: 935–948. [158] Moscat J, Karin M, Díaz-Meco MT. p62 en cáncer: adaptador de señalización más [172] Yuen MF, Chen DS, Dusheiko GM, Janssen HLA, Lau DTY, Locarnini SA, et al. Infección por el virus de la hepatitis B. Nat Rev Dis Primers 2018; 4: 18035. [173] Thrift AP, El-Serag HB, Kanwal F.Epidemiología global y carga de infección por VHC y enfermedad relacionada con el VHC. Nat Rev Gastroenterol Hepatol 2017; 14: 122-132. allá de la autofagia. Cell 2016; 167: 606–609. [174] Lazar C, Uta M, Branza-Nichita N. Modulación de la respuesta de la proteína desplegada [159] Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, et al. La fosforilación de p62 activa la vía Keap1-Nrf2 durante la autofagia selectiva. Mol Cell 2013; 51: 618–631. [160] Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Activación persistente de Nrf2 a través de p62 en células de carcinoma hepatocelular. J Cell Biol 2011; 193: 275–284. [161] Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, et al. La autofagia promueve la invasión de las células del carcinoma hepatocelular mediante la activación de la transición epitelial-mesenquimatosa. Carcinogénesis 2013; 34: 1343–1351. [162] Hu T, Li P, Luo Z, Chen X, Zhang J, Wang C, et al. La cloroquina inhibe el crecimiento de las células del carcinoma hepatocelular in vitro e in vivo. Oncol Rep 2016; 35: 43–49. [163] Wang Y, Zhao H, Wang D, Hao M, Kong C, Zhao X, et al. La inhibición de la autofagia promovió la apoptosis y suprimió el crecimiento del carcinoma hepatocelular tras la exposición fototérmica. J Biomed Nanotechnol 2019; 15: 813–821. [164] Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, et al. La inhibición de la autofagia potencia el efecto antitumoral del inhibidor multiquinasa sorafenib en el carcinoma hepatocelular. Int J Cancer 2012; 131: 548–557. [165] Clarke AJ, Simon AK. Autofagia en la renovación, diferenciación y homeostasis de las células inmunes. Nat Rev Immunol 2019; 19: 170–183. [166] Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. Los sellos distintivos de la inmunoterapia anticancerosa exitosa. Sci Transl Med 2018; 10. por el virus de la hepatitis B humana. Microbiol delantero 2014; 5: 433. [175] Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotipo 1a crecimiento e inducción de autofagia. J Virol 2008; 82: 2241–2249. [176] Rautou PE, Cazals-Hatem D, Feldmann G, Mansouri A, Grodet A, Barcaza S, et al. Cambios en la respuesta autofágica en pacientes con infección crónica por el virus de la hepatitis C. Am J Pathol 2011; 178: 2708–2715. [177] Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, et al. La proteína X del virus de la hepatitis B sensibiliza a las células a la autofagia inducida por inanición mediante la regulación positiva de la expresión de beclin 1. Hepatología 2009; 49: 60–71. [178] Aweya JJ, Mak TM, Lim SG, Tan YJ. La proteína p7 del virus de la hepatitis C induce la muerte celular de manera diferente a la viroporina del virus de la influenza A M2. Virus Res 2013; 172: 24–34. [179] Rios-Ocampo WA, Daemen T, Buist-Homan M, Faber KN, Navas MC, Moshage H. El núcleo del virus de la hepatitis C o la expresión de la proteína NS3 / 4A precondicionan a los hepatocitos contra el estrés oxidativo y el estrés del retículo endoplásmico. Redox Rep 2019; 24: 17-26. [180] Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 y fosfoinositido 3quinasa Vps34 de clase III están implicados en la autofagia inducida por NS4B del virus de la hepatitis C. J Virol 2011; 85: 10561–10571. [167] Soria LR, Brunetti-Pierri N. Focalización de la autofagia para el tratamiento de la hiperamonemia. Autofagia 2018; 14: 1273–1275. [181] Doring T, Zeyen L, Bartusch C, Prange R. El virus de la hepatitis B subvierte el complejo de elongación de autofagia Atg5-12 / 16L1 y no requiere la lipidación de Atg8 / LC3 para la maduración viral. J Virol 2018; 92: e01513 – e1517. [182] Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. La vía autofágica temprana es activada por el virus de la hepatitis B y es necesaria para la replicación del ADN viral. Proc Natl Acad Sci USA 2010; 107: 4383–4388. [168] Eng CH, Yu K, Lucas J, White E, Abraham RT. El amoníaco derivado de la glutaminólisis es un regulador difusible de la autofagia. Sci Signal 2010; 3: ra31. [183] Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K.La eliminación del gen relacionado con la autofagia disminuye la producción de partículas infecciosas del virus de la hepatitis C. Autofagia 2009; 5: 937–945. [169] Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. La autofagia inducida por amoniaco es independiente de las quinasas ULK1 / ULK2. Proc Natl Acad Sci US A 2011; 108: 11121–11126. 196 Revista de Hepatología 2020 vol. 72j 183–196