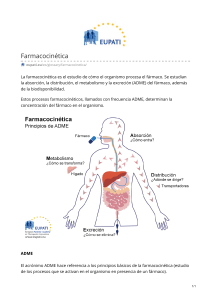
INHIBIDORES DE LA PARED CELULAR PENICILINAS Fármaco Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas Penicilinas naturales • Penicilina G cristalina (penicilina G sódica o potásica) • Penicilina G procaínica • Penicilina G Benzatinica • Fenoximetil penicilina (penicilina V) Las penicilinas están entre los antibióticos con una eficacia más amplia y también entre los fármacos conocidos con una toxicidad menor, pero el aumento de las resistencias ha limitado su uso. Los miembros de esta familia difieren entre sí en las sustituciones en el grupo R unido al residuo del ácido 6aminopenicilánico. La naturaleza de esta cadena lateral influye en el espectro antibacteriano, así como en la Administración: La vía de administración de un antibiótico β-lactámico está determinada por la estabilidad del fármaco en medio ácido y por la gravedad de la infección. a. Vías de administración: Ticarcilina, carbenicilina, piperacilina y las combinaciones de ampicilina con sulbactam, de ticarcilina con ácido clavulánico y de piperacilina con tazobactam se han de administrar por vía i.v. o i.m. La fenoximetilpenicilina, la amoxicilina, la amoxicilina combinada con ácido clavulánico y el éster indanilo de carbenicilina (para el tratamiento de las infecciones del tracto Aquellos microorganismos cuya pared celular carece de peptidoglucano o es impermeable a estos fármacos presentan una resistencia natural a las penicilinas (p. ej., Mycoplasma). La resistencia adquirida a las penicilinas por transferencia de plásmidos se ha convertido en un importante problema clínico, pues un patógeno puede volverse resistente a varios antibióticos al mismo tiempo por adquisición de un plásmido que codifica la resistencia a múltiples fármacos. La Penicilina G COCOS GRAM+ Streptococo pneumoniae Streptococo pyogenes Streptococo viridians Bacilos gran + Bacilo anthracis Corynebacterium diptheriae COCOS GRAM Neisseria gonorrhoae Neisseria meningitidis Anaerobios Clostridium perfringes Espiroquetas Treponema pallidum Treponema pertenue Ampicilina y Amoxcicilina COCOS GRAM + Enterococos BACILOS GRAM+ Las penicilinas se cuentan entre los fármacos más inocuos, y no es necesario controlar sus concentraciones en sangre. Sin embargo, pueden aparecer las reacciones adversas que se describen a continuación Penicilinas antiestafilocóccicas: • Meticilina • Nafcilina • Oxacilina • Dicloxacilina Penicilinas de espectro ampliado: • Ampicilina • Amoxicilina Penicilinas antiseudomonas: • Carbenicilina • Ticarcilina • Piperacilina estabilidad frente a los ácidos gástricos y en la susceptibilidad a las enzimas degradantes bacterianas (βlactamasas). urinario) se encuentran disponibles solamente en formulaciones orales. Otros fármacos son eficaces por las vías i.v., i.m. o v.o. b. Formas de depósito: La bencilpenicilina procaína y la bencilpenicilina benzatina se Administran por vía i.m. y actúan por liberación prolongada. Se absorben lentamente hacia la circulación y persisten a bajas concentraciones durante un largo período. 2. Absorción: La mayoría de las penicilinas se absorben de forma incompleta después de la administración oral y llegan al intestino en cantidades suficientes como para alterar la composición de la flora intestinal. Sin embargo, la amoxicilina se absorbe casi por completo y no es apropiada para el tratamiento de la enteritis por Shigella o Salmonella, pues no se alcanzan concentraciones terapéuticamente eficaces en las criptas intestinales. La absorción de todas las multiplicación de un microorganismo de esta clase aumenta la diseminación de los genes resistentes. Las bacterias pueden adquirir una o más de las propiedades que se describen a continuación mediante la incorporación de un plásmido de resistencia, propiedades que les permiten resistir la acción de los antibióticos βlactámicos. Listeria monocitogenes BACILOS GRAM Eschericha coli Haemophilus influenzae Salmonella typhi Ticarcilina y Pepeacilina BACILOS GRAMEnterobacter spp. Escherichia coli Proteus mirabillis Proteus (Indolpositivo) Haemophilus influenzae Pseudomonas aeruginosa CEFALOSPORINAS Fárma co Farmacodinamia CEFALOSPORINAS Cefalosporinas Farmacocinética Resistencia Muchas cefalosporinas Imipenem y meropenem se administran por vía i.v. Los mecanismos de resistencia bacteriana deben administrarse por vía i.v. o i.m. Debido a su escasa absorción por v.o. Las excepciones a esta norma se exponen en la figura 31.12. Distribución: Todas las cefalosporinas se distribuyen muy bien por los líquidos del organismo. y penetran bien en los tejidos y líquidos corporales, incluido el LCR cuando hay inflamación meníngea. Se excretan por filtración glomerular. El imipenem es escindido por una dehidropeptidasa que se encuentra en el borde en cepillo del túbulo proximal renal. Esta son esencialmente los mismos que los descritos para las Penicilinas. [Nota: aunque no son susceptibles a la hidrólisis por la penicilinasa estafilocócica, las cefalosporinas puedenser sensibles a las β- Administración: d e generación • Cefazolina • Cefadroxilo • Cefalexina • Cefalotina • Cefapirina • Cefradina Cefalosporinas de generación • Cefamandol • Cefronicid 1e ra 2d a Sin Espectro antibacteria no Cefalosporinas de 1ª generación COCOS GRAM + Staphylococ cusaureus Staphylococ cus epidermidis Streptococc us pneumoniae Streptococc us pyogenes Estreptococ os anaeróbicos Reacciones adversas Manifestaciones alérgicas: los pacientes que han presentado una reacción anafiláctica frente a las penicilinas no deben recibir cefalosporinas; hay que evitar o utilizar con precaución en los individuos alérgicos a las penicilinas • Cefoxitina • Cefuroxima • Cefotetan • Cefprozil • Cefaclor • Cefmetazole • Ceforanide • Ceforinida • Loracarbef Cefalosporinas de generación: • Ceftazidima • Cefotaxima • Ceftriaxona • Cefixima • Cefeperazone • Moxalactan • Cefpodoxima • Ceftibuten Cefalosporinas de generación • Cefepina • Cefpiroma Generación avanzada • Ceftarolina 3ra 4ta embargo, las concentraciones terapéuticas adecuadas en el LCR, independientemente de la inflamación, sólo se alcanzan con las cefalosporinas de tercera generación. Por ejemplo, la ceftriaxona o la cefotaxima son eficaces en el tratamiento de la meningitis neonatal e infantil producida por H. influenzae. La cefazolina se aplica en una dosis profiláctica única antes de la cirugía, por su semivida de 1,8 h y su actividad frente a S. aureus productor de penicilinasa. Si la intervención dura más de 3 h puede ser necesario administrar dosis adicionales intraoperatorias. La cefazolina es eficaz en la mayoría de procedimientos quirúrgicos, incluida la cirugía ortopédica, por su capacidad de penetración intraósea. enzima forma un metabolito inactivo que puede ser nefrotóxico. La combinación de imipenem con cilastatina protege el fármaco original y, por lo tanto, evita la formación del metabolito tóxico; esto hace posible la utilización del fármaco en el tratamiento de las infecciones urinarias. El meropenem no se metaboliza. El ertapenem puede administrarse por vía i.v. o i.m. lactamasas de espectro ampliado.] BACILOS GRAMEscherichia coli Klebslella pneumoniae Proteus mirabilis Cefalosporinas de 2ª generación COCOS GRAM+ Staphylococcus aureus Streptococcus pneumoniae Streptococcus pyogenes Estreptococos anaeróbicos COCOS GRAMNelsseria gonorrhoeae BACILOS GRAMEnterobacter aerogenes Escherichia coli Haemophilus influenzae Klebslella pneumoniae Prteus mirabilis Cefalosporinas de 3ª generación COCOS GRAM+ Streptococcus pneumoniae Todas las cefalosporinas atraviesan la placenta. Destino: La biotransformación de las cefalosporinas en el huésped no tiene importancia clínica. La eliminación se produce por secreción tubular y/o filtración glomerular. Hay que ajustar las dosis en caso de insuficiencia renal grave para evitar la acumulación y los efectos tóxicos. La ceftriaxona se excreta a través de la bilis hacia las heces. Por lo tanto, se emplea con frecuencia en los pacientes con insuficiencia renal. Strptococcus pyogenes Estreptococos anaeróbicos COCOS GRAMNelsseria gonorrhoeae BACILOS GRAMEnterobacter aerogenes Escherichia coli Haemophilus influenzae Klebsiella pneumoniae Proteus mirabilis Pseudomonas aeruginosa CARBAPENEMICOS Fármaco Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas Carbapenemicos Diropenem Ertapenem Impemem/cilastatina Meropenem Los carbapenémicos son antibióticos βlactámicos sintéticos cuya estructura difiere de la de las penicilinas en el átomo de azufre del anillo de tiazolidina , que ha sido externalizado y sustituido por un átomo de carbono . El imipenem, el meropenem y el ertapenem son los únicos fármacos de este grupo disponibles en la actualidad. El imipenem se combina con cilastatina para protegerlo frente al metabolismo por la dehidropeptidasa renal. Imipenem y meropenem se administran por vía i.v. y penetran bien en los tejidos y líquidos corporales, incluido el LCR cuando hay inflamación meníngea. Se excretan por filtración glomerular. El imipenem es escindido por una dehidropeptidasa que se encuentra en el borde en cepillo del túbulo proximal renal. Esta enzima forma un metabolito inactivo que puede ser nefrotóxico. La combinación de imipenem con cilastatina protege el fármaco original y, por lo tanto, evita la formación del metabolito tóxico; esto hace posible la utilización del fármaco en el tratamiento de las infecciones urinarias. El meropenem no se metaboliza. El ertapenem puede administrarse por vía i.v. o i.m. [Nota: en los pacientes con insuficiencia renal deben ajustarse las dosis de estos fármacos.] Algunas cepas de sudomonas son resistentes, y también aparición de cepas resistentes de P. aeruginosa en tratamientos COCO GRAM+ Staphylococcus aureus Staphylococcus epidermidis Enterococcus fecalis Estreptococos de los grupos A, B, C Streptococcus pneumoniae BACILOS GRAM + Listeria monocytones COCOS GRAMNeisseria gonorrhoeae Neisseria meningitidis Bacilos GRAM – Acinetobacter spp. Citobacter spp ENterobacter spp Escherichia coli Gardnerella vaginalis Haemophilus influenzae Klebsiella spp Pseudomonas aeruginosa Salmonella spp Serratia spp Anaerobios Clostridium spp Peptococcus spp La combinación imipenem/cilastatina puede provocar náuseas, vómitos y diarrea. La eosinofilia y la neutropenia son menos frecuentes que con el resto de β-lactámicos. Los niveles elevados de imipenem pueden producir convulsiones, mientras quela incidencia de este efecto adverso es menor con el meropenem. En larealización de estudios con animales, el doripenem no ha demostrado ningún potencial para causar convulsiones. Peptostreptococcus spp Propionibacterium spp Bacteroides spp Fusobacterium spp MONOBACTAMICOS Fármaco Monobactámicos Aztreonam Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas La peculiaridad de los monobactámicos, que también alteran la síntesis de la pared bacteriana, es que el anillo B-lactámico no está fusionado con otro anillo Administración: Via i.v o i.m. Excreción: orina EL aztreoman es resistente a la acción de las B-lactamasas Actividad antibacteriana principalmente contra las ENTEROBACTERIÁCEAS, frente a los BACILOS AEROBIOS GRAM NEGATIVO - P. aeruginosa El aztreoman es relativamanete atóxico, aunque puede producir: Fleblitis Exantemas cutáneos Ocasionalmente, alteración de las pruebas funcionales hepáticas INHIBIDORES DE LA B-LACTAMASA Fármaco Inhibidores de la B-lactamasa Ácido clavulanico Sulbactam Tazobactam Farmacodinamia Se unen a las Blactamasas y las inactivan, protegiendo así a los antibióticos que normalmente son Farmacocinética Resistencia Espectro antibacteriano Carecen de actividad antibacteriana Reacciones adversas sustratos de estas enzimas VANCOMICINA Fármaco Vancomicina Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas La vancomicina es un glucopéptido tricíclico que ha adquirido una creciente importancia por su eficacia frente a múltiples microorganismos resistentes a los fármacos, como los SARM y los enterococos. En la actualidad existen problemas por la aparición de resistencia a la vancomicina entre estos microorganismos. [Nota: la bacitracina es una mezcla de polipéptidos que también inhibe la síntesis de la pared bacteriana. Es activa frente a una amplia variedad de gram positivos, pero su uso está limitado a la aplicación tópica a causa de su La infusión i.v. lenta (60-90 min) se emplea para el tratamiento o la profilaxis de las infecciones sistémicas. Como la vancomicina no se absorbe tras la administración oral, esta vía se emplea sólo para el tratamiento de la colitis por C. difficile inducida por antibióticos, en los casos en que ha fracasado el metronidazol. La inflamación permite que el fármaco penetre en las meninges. Sin embargo, a menudo es necesario combinar la vancomicina con otros antibióticos, como la ceftriaxona, para obtener efectos sinérgicos en el tratamiento de la meningitis. El metabolismo del fármaco es mínimo, y el 90-100% se excreta por filtración glomerular (fig. La resistencia a la vancomicina puede deberse a cambios en la permeabilidad al fármaco mediados por los plásmidos o a una reducción de la unión de la vancomicina con las moléculas del receptor. [Nota: un ejemplo de esto último se halla en el reemplazamiento de un D-Ala por un D-lactato en los microorganismos resistentes.] COCOS GRAM+ Staphylococcus aureus Staphylococcus epidermidis Enterococos de los grupos A, B, C Streptococcus pneumoniae Enterococcus faecalis BACILOS GRAM+ Listeria monocytogenes Corynebacterium jelkelum ANAEROBIOS Clostridium spp OTROS Actimonomyces spp Los efectos adversos de la vancomicina son un problema importante y consisten en fiebre, escalofríos y/o flebitis en el lugar de la infusión. Si el ritmo de la infusión es rápido, se produce enrojecimiento («síndrome del hombre rojo») y choque por liberación de histamina. En caso de reacción infusional hay que reducir la velocidad de administración para que dure unas 2 h, o bien aumentar el volumen de dilución o pretratar con un antihistamínico 1 h antes de la infusión. Además, las reacciones pueden tratarse con antihistamínicos y esteroides .Esta reacción no es una potencial nefrotóxico con el empleo sistémico.] 31.17). [Nota: en la insuficiencia renal deben ajustarse las dosis, pues de lo contrario se acumula el fármaco. La semivida normal es de 6 a 10 h, en comparación con las másde 200 h en caso de enfermedad renal en fase avanzada.] alergia, y los clínicos deben tener cautela de no confundirla con verdadera hipersensibilidad. DAPTOMISINA Fármaco Daptomicina Farmacodinamia Farmacocinética La daptomicina es un antibiótico lipopeptídico cíclico que constituye una alternativa al empleo de otros agentes, como la linezolida y la combinación quinupristina/dalfopristin a, para el tratamiento de las infecciones producidas por microorganismos gram positivos resistentes, incluidos los SARM y enterococos resistentes a vancomicina (ERV). La daptomicina está unida en un 90-95% a las proteínas y no se metaboliza en el hígado; sin embargo, en los pacientes con afectación renal deben ajustarse las dosis (depuración de creatinina inferior a 30 ml/min). En las infecciones de la piel y los tejidos blandos, se administra una infusión de 4 mg/kg/día i.v. durante 30 min. Resistencia Espectro antibacteriano COCOS GRAM+ Staphylococcus aureus (SARM y SASM) Enterococo faecalis Enterococo faecium Streptococos pneumoniae Streptococos pyogenes BACILOS GRAM + Corynebacterium jeikeium Reacciones adversas Los efectos adversos más frecuentes observados en los ensayos clínicos consisten en estreñimiento, náuseas, cefalea e insomnio. Pueden aumentar las cifras de transaminasas hepáticas y de creatínfosfocinasas, razón por la que es aconsejable realizar controles semanales. TELAVANCINA Fármaco Telavancina Farmacodinamia Farmacocinética La telavancina es un antibiótico lipoglucopéptido semisintético que constituye un derivado sintético de la vancomicina. Es una alternativa a vancomicina, daptomicina, linezolida y quinupristina/dalfopristina para tratar infecciones cutáneas y de la estructura cutánea complicadas producidas por microorganismos gram positivos resistentes, incluido SARM. No se sabe si la telavancina experimenta metabolismo hepático, pero tiene semivida de 7 a 9 h. Se administra a razón de 10 mg/kg en una infusión de 60 min cada 24 h (fig. 31.20). En pacientes con depuración de creatinina entre 30 y 50 ml/min, la dosis se reduce a 7.5 mg/kg cada 24 h. En pacientes con depuración de creatinina de 10 a 29 ml/min, la dosis recomendada es de 10 mg/kg con intervalo de 48 h. Por tanto, el funcionamiento renal debe vigilarse durante el tratamiento, pero no es necesario vigilar la concentración sérica de telavancina. Resistencia Espectro antibacteriano Reacciones adversas COCOS GRAM+ Straphylococcus auereus (SARM y SASM) Enterococcus faecalis Streetococcus pneumoniae (resistente a penicilina) Streptococcus pyogenes Las reacciones adversas más comunes informadas con la telavancina han incluido perversiones del sentido del gusto, náuseas, vómito, insomnio y orina espumosa. No se recomienda durante el embarazo, debido a resultados adversos del desarrollo observados en animales. En Estados Unidos el empaque tiene un recuadro en el que se advierte a las mujeres en edad reproductiva que se realicen una prueba de embarazo antes de usar el medicamento. Dado que la telavancina podría prolongar el intervalo QTc, debe evitarse su uso en pacientes con el antecedente de prolongación de dicho intervalo, insuficiencia cardíaca descompensada, hipertrofia grave del ventrículo izquierdo o uso de otros fármacos que podrían prolongar el intervalo QTc. POLIMIXINAS Fármaco Polimixinas Farmacodinamia Es un inhibidor de la pared celular bacteriana Farmacocinética Solo dos formas de polimixina tienen uso clínico, la polimixina B esta disponible en presentación paraenteral, oftálmica, otica y topica Lacolistina (polimicina E) solo puede obtenerse como promedicamento, el colistimeato sódico que se administra por vía IV o inhalado con nebulizador Resistencia Espectro antibacteriano BACILOS GRAMEscherichia coli Klepsiella spp Enterobacter spp Ascinetobacter spp Pseudomona auruginosa Reacciones adversas El empleo de estos medicamentos se ah limitado por mucho tiempo debido al alto riesgo de nefrotoxicidad y neurotoxicidad ejemplo (lenguaje, confuso, debilidad muscular) cuando se administra por via sistémica. Sin embargo con el aumento de resistencia entre gran-, su uso a resurgido y ahora son comunes en el tratamiento de salvamento con infecciones resistentes a múltiples medicamentos. INHIBIDORES DE LA SINTESIS DE LAS PROTEINAS TETRACICLINAS • • • • Fármaco Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas Demeclociclina Doxiciclina Minociclina Las tetraciclinas son un grupo de compuestos estrechamente relacionados que, como su nombre indica, poseen cuatro anillos fusionados con un sistema de dobles enlaces conjugados. Las diversas sustituciones en estos anillos explican las diferencias en la farmacocinética de cada fármaco, que se asocian a pequeñas diferencias en su eficacia clínica. Absorción: Todas las tetraciclinas se absorben adecuada pero incompletamente por v.o. Sin embargo, si se toman estos fármacos con alimentos lácteos se forman quelatos no absorbibles de las tetraciclinas con los iones de calcio y disminuye la absorción de éstas. También se forman quelatos no absorbibles con otros cationes divalentes y trivalentes (p. ej., los que se hallan en los antiácidos con magnesio y aluminio y en los preparados La amplia resistencia existente a las tetraciclinas limita su uso clínico. La resistencia natural más frecuente, o factor «R», confiere al microorganismo una incapacidad para acumular el fármaco, que se concreta en un flujo activo de salida del fármaco dependiente de Mg++, mediado por la proteína de resistencia BACILOS GRAM + Bacilos anthracis BACILOS GRAM – Brucella spp Vibrio cholerae Yersinia pestis ANAEROBIOS Clostridium tetani Clostridium perfringes ESPIROQUETAS Borerelia burgdorferi Leptospira Interrogans Molestias gástricas: suelen deberse a una irratacion de la mucosa del estomago y a menudo son responsables de la falta de colaboración del paciente Efecto sobre los tejidos calcificados: En el periodo de crecimiento y calcificación de de la infancia estos Tetraciclina con hierro). [Nota: esto puede ser un problema si el paciente se automedica con antiácidos para las molestias gástricas causadas por la ingestión de tetraciclinas. La doxiciclina y la minociclina se absorben casi totalmente trassu administración oral. En la actualidad, la doxiciclina es la tetraciclina de elección para la administración parenteral. TetA codificada por los plásmidos. Otros mecanismos menos importantes de resistencia bacteriana a las tetraciclinas incluyen la inactivación enzimática del fármaco y la producción de proteínas bacterianas que impiden la unión de las tetraciclinas al ribosoma. Cualquier microorganismo que sea resistente a una tetraciclina lo es a todas. La mayoría de los estafilococos productores de penicilinasa son actualmente resistentes a las tetraciclinas. fármacos se depositan en el hueso y en la dentición primaria y provovan alteraciones en los dientes e hipoplasia, como interruocion en el crecimiento Toxicidad hepática letal: se ven en embarazadas que reciben altas dosis de tetraciclinas Fototoxicidad: se exponen a la luz solar o a los rayos ultravioleta se produce fototoxicidad Trastornos vestibulares: principalmente con la minociclina Seudomotor cerebral: Hipertensión intracraneal benigna, acompañada de cefalea y visión borrosa Sobreinfecciones GLICILCICLINAS Fármaco GLICILCICLINAS Tigeciclina Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas La tigeciclina es el primer miembro disponible de una nueva clase de antibacterianos denominados glicilciclinas. La tigeciclina, un derivado de la minociclina, es similar estructuralmente a las tetraciclinas y posee una acción de amplio espectro contra los patógenos Gram positivos resistentes a múltiples fármacos, así como frente a algunos microorganismos Gram negativos y anaerobios. La tigeciclina está indicada en el tratamiento de las infecciones complicadas de la piel y los tejidos blandos, y también en las infecciones intraabdominales difíciles. Después de una infusión i.v. durante 30 a 60 min cada 12 h, la tigeciclina se distribuye por todo el plasma y los tejidos corporales. No experimenta una metabolismo hepático importante y se elimina principalmente por vía biliar/fecal. No es necesario ajustar las dosis en los pacientes con trastornos renales, pero sí en caso de disfunción hepática grave. La tigeciclina se desarrolló para hacer frente a la reciente aparición de microorganismos resistentes al grupo de las tetraciclinas, que utilizan los mecanismos de bomba de salida y protección ribosómica para obtener resistencia. COCOS GRAM+ Staphylococcus (SARM) Enterococos resistentes a la vancomicina Streptococos resistentes a múltiples medicamentos Estreptococo de los grupos A, B, C BACILOS GRAM Productora de Blactamsa de espectro extentdido Ascinetobacter baumanni y muchos microorganismos anaerobios No es active contra morganella Proteus La tigeciclina es bien tolerada, y sus principales efectos adversos son similares a los producidos por el grupo de las tetraciclinas. En los ensayos clínicos, los efectos adversos observados con más frecuencia fueron náuseas y vómitos. Otros efectos secundarios similares a los producidos por las tetraciclinas son: fotosensibilidad, seudotumor cerebral, coloración de los dientes si se administra durante el desarrollo dentario, y daño fetal. AMINOGLUCOSIDOS Fármaco AMINOGLUCOSIDOS Amikacina Estreptomicina Gentamicina Neomicina Tobramicina Farmacodinamia Farmacocinética Resistencia Los antibióticos aminoglucósidos habían sido la base del tratamiento de las infecciones graves por bacilos aerobios Gram negativos, pero los importantes efectos tóxicos asociados justifican que hayan sido reemplazados hasta cierto punto por otros antibióticos más seguros, como las cefalosporinas de tercera y cuarta generación, las fluoroquinolonas y los carbapenémicos. En inglés, los aminoglucósidos derivados de Streptomyces tienen Administración: Todos (excepto la neomicina) deben administrarse por vía parenteral para alcanzar unos niveles séricos adecuados Distribución: Su potencial nefrotóxico y ototóxico se explica por qué las concentraciones elevadas se almacenan en la corteza renal y en la endolinfa y perilinfa del odio interno. Todos los aminoglucósidos atraviesan la barrera placentaria y pueden acumularse en el plasma fetal y en el liquido amniótico Destino: todos se excretan rápidamente por la orina, sobre tofo por filtración glomerular Se produce resistencia por: Disminución de la captación del fármaco en caso de ausencia del sistema de transporte para los aminoglucósidos, dependiente de oxígeno, o de los canales de las porinas Síntesis de enzimas (ej: acetiltransferasas) asociadas a plásmidos que modifican e inactivan los antibióticos aminoglucósidos. Espectro antibacteriano COCOS GRAM + Enterococcos spp (Gentamicina + ampicilina) Sterptococcus agalactiae (Gentamicina + ampicilina) BACILOS GRAMBrucella spp Farcisella Klebsiaella Pseudommona aeruginosa Yersinia pesti Reacciones adversas Ototoxicidad Nefrotoxicidad Parálisis Exantema cutáneo Reacciones alérgicas dermatitis de contacto a la aplicación tópica de la neomicina sufijos con -mycin, mientras que los derivados de Micromonospora acaban en -micin. Los términos «aminoglucósido» y «aminociclitol» derivan de su estructura: dos aminoazúcares unidos por un enlace glucosídico a un núcleo hexosa central (aminociclitol). Su naturaleza policatiónica impide que atraviesen fácilmente las membranas hísticas. Se cree que todos los miembros de esta familia inhiben la síntesis de proteínas bacterianas por el mecanismo descrito para la estreptomicina, como se explica a continuación. MACROLIDOS/CETOLIDOS Fármaco MACROLIDOS/CETOLIDOS Azitromicina Claritromicina Eritromicina Farmacodinamia Farmacocinética Resistencia Los macrólidos son un grupo de antibióticos con estructura de lactona macrocíclica y uno o más desoxiazúcares añadidos. Administración: (V.O) Eritromicina (V.O) La claritromicina, la azitromicina y la telitromicina son estables en el medio acido La resistencia a la eritromicina se está convirtiendo en un problema clínico importante. Por Espectro antibacteriano COCOS GRAM+ Stapphylococcus aureus Streptococos pneumoniae Reacciones adversas Molestias epigástricas Ictericia colestásica Ototoxicidad Contraindicaciones: Pacientes von disfusion Telitromicina La eritromicina fue el primer fármaco de esta clase con aplicación clínica, bien comofármaco de elección o como alternativa a la penicilina en individuos alérgicos a los antibióticos βlactámicos. Los miembros más recientes de esta familia, claritromicina (una forma metilada de eritromicina) y azitromicina (con un anillo lactona más grande), tienen algunas características en común con la eritromicina, además de otras que mejoran las de ésta. La telitromicina, un derivado semisintético de la eritromicina, es el primer antibacteriano «cetólido» que se ha autorizado, y en la actualidad se emplea clínicamente. Aunque los cetólidos y los macrólidos tienen una cobertura antimicrobiana muy similar, los primeros son activos frente a muchas cepas Gram positivas resistentes a los segundos. del estomago y se absorben con facilidad Azitromicina (I.V) EL empleo de la eritromicina i.v se asocia a una elevada incidencia de tromboflebitis Distribucion: Eritromicina se distribuye por todos los líquidos corporales, excepto el LCR Los cuatro fármacos se concentran en el hígado La claritromicina, la azitromicina y la telitromicina se distribuyen por los tejidos Metabolismo: la eritromicina y la telitromicina se metabolizan estensamente e inhiben la oxidación de ciertos fármacos a través de su interacción con el sistema del citocromo P450 La claritromicina interfiere en el metabolismo de fármacos como la teofilina y carbamazepina La claritromicina se oxida derivado 14-hidroxi Excrecion: La eritromicina y la azitromicina se concentran principalmente en la bilis y se excreta por la misma La claritromicina se excreta por el riñón y el hígado ejemplo, la mayoría de las cepas estafilocócicas intrahospitalarias son resistentes a este fármaco. Se han identificado varios mecanismos de resistencia: 1) la incapacidad del microorganismo para captar el antibiótico o la presencia de una bomba con flujo de salida; ambos limitan la cantidad de fármaco intracelular; 2) disminución de la afinidad de la subunidad ribosómica 50S por el antibiótico por metilación de una adenina en la subunidad 23S del ARN ribosómico bacteriano, y 3) presencia de una eritromicinesterasa asociada a los plásmidos. La claritromicina y la azitromicina presentan resistencia cruzada con la eritromicina, pero la telitromicina puede ser eficaz contra los microorganismos Strptococos Pyogenes BACILOS GRAM+ Corubacterium diptheriae COCOS GRAMNeisseria gonorrhoae Moraxella catarrhalis BACILOS GRAM – Bordetella pertussis Campylobacter jejuni Haemophilus influenzae Legionella pneumoniae ESPIROQUETA Treponema pallidum MICOPLASMA Mycoplasma pneumoniae Ureoplasma urealyticum CLAMDIOS Chlamidia pneumoniae Chlamidia psittaci Chlamidia trachomatis hepática hay que emplear con precacaucion la eritromicina, la telitromicina y la azitromicina, se observan casos de hepatotoxicidad Interacciones: La eritromicina, la telitromicina y la claritromicina inhiben el metabolismo hepático de algunos fármacos resistentes macrólidos. a los MACROCICLICOS Fármaco MACROCICLICOS Fidaxomicina Farmacodinamia Farmacocinética Antibiótico macrocíclico con una estructura similar a los macrólidos, tiene un mecanismo de acción singular actúa sobre la subunidad sigma del ARN polimerasa con lo que altera la transcripción bacteriana, termina la síntesis de la proteína y produce la muerte celular en microorganismos Distribcion: V.O Absorción: Mínima y la mayor parte pertenece en el tracto gastrointestinal esto es ideal para el tratamiento de la infección por dostridiun defficile que tiene lugar en el intestino Resistencia No hay registro Espectro antibacteriano Reacciones adversas Se limita a aeróbicos y anaeróbicos gran+ Posee actividad contra estafilococos y enterococos se utiliza principalmente por su actividad bacteriana contra la clostridium difficile Nauceas Vomito Dolor abdominal Angioedema Disnea Prurito Fidaxomicina debe ser administrada con precaución en pacientes con alergia a macrólidos CLORANFENICOL Fármaco CLORANFENICOL Farmacodinamia Farmacocinética Resistencia Se une a la subunidad ribosómica 50S bacteriana e inhibe la síntesis de proteínas en la reacción de la peptidiltransferasa Puede administrarse por via i,v o v.o Su naturaleza lipófila permite su absorción completa por v.o y se Presencia de un factor R que codifica una acetilcoenzima A transferasa que inactiva el cloranfenicol. Espectro antibacteriano Reacciones adversas Antibiótico de amplio Anemias espectro, no solo con las Síndrome del bacterias sino también lactante gris frente a otros Interacciones microorganismos como distribuye por todo el organismo, penetra el LCR. Inhibe las oxidasas hepáticas de función mixta Su excreción depende se su conversión hepática en un glucoronido, que luego se secreta en el tubular renal Otro es la incapacidad del antibiótico para penetrar al patógeno las rickettisias, La actividad del cloranfenicol contra los anaerobios es excelente Es bactericida o bacteriostático Resistencia Espectro antibacteriano Reacciones adversas Microorganismo gran + incluidos el SARM, estreptococos y bacterias anaeróbicas Exantema cutaneo Diarrea (efecto más grave) Colitis pseudomembranosa producida por sobrecrecimiento de C. CLINDAMICINA Fármaco CLINDAMICINA Farmacodinamia Farmacocinética El mecanismo de acción es el mismo que el de la eritromicina, se une de modo irreversible a un lugar de la subunidad 50S del ribosoma bacteriano e inhibe la síntesis de las proteínas Administración: I.v y v.o La administración oral está limitada por la intolerancia gástrica Presenta una amplia distribución por todos los líquidos corporales, a excepción del LCR Penetra el tejido óseo, aunque no haya inflamación. Presenta un amplio metabolismo oxidativo hasta la formación de productos inactivos, y se excreta en la bilis o en la orina por filtración glomerular Es igual a los de la eritromicina QUINUPRISTINA/ DALFOPRISTINA difficile, que elabora toxinas necrosantes y potencialmente letal Fármaco QUINUPRISTINA/ DALFOPRISTINA Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Cada componente se une a un lugar diferente en la subunidad 50S del ribosoma bacteriano para formar un complejo terciario estable Se inyecta por via i.v en suero glucosado al 5% La mayor parte de los fármacos originales y sus metabolitos se eliminan a través del hígado y se excretan por la bilis a las heces La excreción urinaria es secundaria Los procesos enzimáticos suelen ser el origen de la resistencia a estos fármacos. La presencia de una enzima ribosómica que metila el blanco ARN 23S del ribosoma bacteriano interfieren en la unión de la quinupristina Combinado es principalmente activo contra los COCOS GRAM +, incluidos los resistentes a otros antibióticos. Se utiliza principalmente en el tratamiento de la infección por E. faecium, incluidad las cepas ERV Reacciones adversas Irritación venosa Artralgias y mialgias Hiperbilirrubinemia Interacciones LINEZOLINA Fármaco LINEZOLINA Farmacodinamia Farmacocinética Resistencia Inhibe la formación del complejo de iniciación 70S y por consiguiente la síntesis de las proteínas bacterianas. Se une a la subunidad 50S en un punto cercano a la interfase con la subunidad 30S Se absorbe completamente tras su administración oral (también en I.v) Se distribuye ampliamente por todo el organismo, con volumen de distribución de 40 a 50L Se excreta por las vías renal y extra renal y se eliminan por la orina La disminución de la unión al sitio diana confiere resistencia a los patógenos. No se produce resistencia cruzada con otros antibióticos Espectro antibacteriano COCOS GRAM+ Staphylococcus epidermies Staphylococcus haemoliticus Enterococo feacalis (resistente a la vancomicina) Enterococo faecium (resistentes a la vancomicina) Reacciones adversas Molestias digestivas, náuseas y diarrea Cefalea Exantemas Trombocitopenia en cerca del 2% de los pacientes Streptococos pneumoniae (resistencia a la penicilina) Streptococos del grupo viridns BACILOS GRAMListeria monocitogenes Corybacterium spp ANAEROBIOS Clstridium perfringes OTROS Micobacterium tuberculosis Fármaco FLUOROQUINOLONAS PRIMERA GENERACIÓN Ácido nalidíxico NEGGRAM SEGUNDA GENERACIÓN Ciprofloxacina Norfloxacina Ofloxacina TERCERA GENERACIÓN Levofloxacina CUARTA GENERACIÓN Moxifloxacina INHIBIDORES DE LA SÍNTESIS DE FOLATO Mafenida Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas Penetran en las bacterias por difusión pasiva a treves de los canales proteicos acuosos (porinas) en la membrana externa. Una vez dentro de la célula, inhiben la replicación del ADN bacteriano interfiriendo en la acción de la ADN girasa (topoisomerasa II) y la topoisomerasa IV durante el crecimiento y la reproducción bacterianas. ABSORCIÓN: Del 35 al 70% de la norfloxacina que se administra por v.o. se absorbe. Las ciprofloxacinas, levofloxacina y ofloxacina se administran por vía i.v. DESTINO: Se distribuyen bien en todos los líquidos y tejidos corporales; en el tejido óseo, la orina, el riñón y el tejido prostático se alcanzan Cepas resistentes de SARM, Pseudomonas, estafilococos negativos a coagulasa y enterococos como consecuencia de mutaciones cromosómicas. Resistencias cruzadas: Alteración del blanco: mutaciones en el ADN girasa bacteriana se han asociado con una disminución de la afinidad fluoroquinolonas PRIMERA GENERACION BACILLOS GRAMLas quinolonas de primera generación menos usadas en la actualidad tienen actividad moderada contra Gram -. Alcanzan concentraciones séricas mínimas y se limitan al tratamiento de infecciones no complicadas de vías Aparato digestivo: Los efectos adversos más comunes de las fluoroquinolonas son náuseas, vómitos y diarrea, que se observan en el 3-6% de los pacientes Problemas del sistema nervioso central: Los efectos sobre el sistema nervisoso central urinarias SEGUNDA GENERACION Sulfadiazina de plata Sulfasalazina Sulfisoxazol INHIBIDORES DE LA REDUCCIÓN DE FOLATO Pirimetamina Trimetoprima COMBINACIÓN DE INHIBIDORES DE LA SÍNTESIS Y REDUCCIÓN DE FOLATO Cotrimoxazol (trimetoprima+sulfametoxazol ) ANTISÉPTICOS DEL TRACTO URINARIO Metenamina Nitrofurantoína Cuando la quinolona se une a la enzima y al ADN, se forma un complejo ternario que inhibe el paso del nuevo cierre y producir la muerte celular por inducción de la escisión del ADN concentraciones elevadas, pero no en el líquido prostático. Las concentraciones en el pulmón superan a las del suero. La penetración en el LCR es escasa, a excepción de la ofloxacina. Se acumulan en los macrófagos y en los leucocitos polimorfonucleares. Se eliminan por vía renal, por lo tanto, la dosis se debe ajustar cuando se produzcan cambios de la función renal. La Moxifloxacina se excreta principalmente por el hígado, y no se requiere un ajuste de dosis con la disminución del funcionamiento renal. Disminución de la acumulación: la disminución de la concentración de los fármacos en el interior de la célula bacteriana obedece a dos mecanismos. Uno de ellos es el decenso en el numero de porinas proteicas en la membrana externa de las células resistentes que dificulta el acceso de los fármacos a las topoisomerasas intracelulares. El otro mecanismo se asocia con un sistema de bomba de salida, dependiente de energía COCOS GRAM+ BACILLOS GRAM- MICOPLASMAS CLAMIDIAS Las fluroquiniolonas de segunda generación tienen actividad ampliada contra Gram negativo y alguna actividad contra Gram positivos y microorganismos atípicos, como Mycoplasma pneumoniae y Chiamydia pneumoniae TERCERA GENERACION COCOS GRAM+ BACILOS GRAMMICOPLASMAS CLAMIDIAS Las fluroquinolonas de tercera generación conserva actividad ampliada contra Gram negativo y mejor actividad contra microorganismos atípicos y bacterias Gram + especificas CUARTA GENERACION COCOS GRAM+ BACILLOS GRAM+ BACILLOS GRAMANAEROBIOS Las fluroquinolonas de cuarta generación tienen mejor cobertura contra Gram +, conservan actividad contra Gram – y son las cefalesas y los mareos o el aturdimiento, Fototoxicidad: A los pacientes que toman fluroquinolonas se les aconseja que eviten exposición excesiva al sol y que se apliquen filtros solares; es posible que estos filtros no consigan una protección completa y al primer signo de fototoxicidad es aconsejable suspender el fármaco Problemas del tejido conjuntivo: Deben evitarse las fluoroquinolonas en el embarazo, en las madres lactantes y en los niños menores de 18 años, ya que se observan erosiones del cartílago articular. Contraindicaciones: la moxifloxacina puede prolongar el ganan en cobertura contra anaerobios intervalo QT y, no debe usarse en los pacientes predispuestos a las arritmias o que toman antirritmicos Interacciones farmacológicas: Antes se ha comentado el efecto de los antiácidos y de los cationes sobre la absorción de estos fármacos SULFAMIDAS Fármaco SULFAMIDAS Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas Las sulfamidas que se utilizan actualmente son análogos sintéticos dl PABA. Por su similitud estructural con el PABA, las sulfamidas compiten con este sustrato por la enzima bacteriana dihidropteroato sintetasa, inhibiendo así el ácido dihidrofolico bacteriano y Administración: V.O Se absorben bien en el intestino delgado. Las sulfamidas i.v se reservan generalmente para los pacientes que no pueden percibirlas por v.o Distribución: se unen a la albumina serica en la circulación, donde el Solo son sensibles a las sulfamidas los microorganismos que cubren sus necesidades de folato sintetizándolo de novo. La resistencia adquirida o los fármacos sulfamidicos puede surgir a partir de la transferencia de plásmidos Son activos frente a determinadas enterobacterias en el tracto urinario y frente a Nocardia. En combinación con la pirimetamina, inhibidora del dihidrofolato reductasa, es tratamiento de elección para la toxoplasmosis y el Cristaluria Hipersensibilidad Trastornos hematopoyéticos Ictericia nuclear Potenciación de fármacos: Desplazamiento de sus lugres de la unión la formación de sus cofactores esenciales. grado de unión depende del pKa de cada fármaco Se distribuyen por toda el agua corporal y penetran bien en el LCR incluso sin inflamación. Pueden atravesar la barrera placentaria y penetrar en los tejidos fetales Metabolismo: se acetilan principalmente en el hígado Excreción: Son eliminadas por filtración glomerular o por mutaciones aleatorios paludismo resistente a cloroquina en la albumina serica puede provocar la potenciación transitoria del efecto hipoglucemiante de la tolbutamida o del efecto anticoagulante de la warfina Contraindicaciones: por la inctericia nuclear, no se debe administrarse a los recién nacidos y lactantes menores de 2 años TRIMETOPRIMA Fármaco TRIMETOPRIMA Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Forma activa del folato es el derivado tetrahidro, que se forma por reducción del ácido dehidrofolico por la dehidrofolato reductasa. Esta reacción es inhibida por la trimetoprima, que disminuye la disponibilidad de las coenzimas tetrahidrofolato que se requieren para la síntesis Forma activa del folato es el derivado tetrahidro, que se forma por reducción del ácido dehidrofolico por la dehidrofolato reductasa. Esta reacción es inhibida por la trimetoprima, que También penetra el liquido cefalorraquideo Se elimina sin cambios a través de la orina En las bacterias Gram-, se debe a la presencia de una dihidrofolato reductasa alterada, que tiene menor afinidad por la trimetoprima. La sobreproducción de la enzima también puede originar resistencia por disminución de las permeabilidades al fármaco Es similar al del sulfametoxazol. El primero es de 20 a 50 veces mas potente que la sulfamida. Puede utilizarse para tratamientos de las ITU agudas, como la prostatitis bacteriana y la vaginitis Reacciones adversas Puede producir los efectos de ácido fólico, que consisten en anemia megaloblástica, leucocitopenia y granulocitopenia de purina, pirimidina y aminoácidos TRIMETOPRIMA-SULFAMETOXAZOL Fármaco TRIMETOPRIMASULFAMETOXAZOL Farmacodinamia La actividad antimicrobiana sinérgica de esta combinación se debe a que inhibe dos pasos secuenciales en la síntesis del acido tetrahidrofólico. Sulfametoxazol inhibe la incorporación del PABA a los precursores del acido tehidrofólico y la trimetoprima evita la reducción del dehidrofolato a tetrahidrofolato Farmacocinética La trimetoprima es más liposoluble que el sulfametoxazol y tiene un volumen de distribución mayor Administración V.O I.V con pacientes con neumonía Resistencia Espectro antibacteriano Es menos frecuente que la de cualquiera de los dos fármacos por separado, ya que se produzca es necesario que los patogenis sean resistentes simultáneamente a ambos agentes Sinergia entra la trimetoprima y el sulfametoxazol puede inhibir el crecimiento de Escherichia coli BACILLOS GRAM+ Listeria monocytogenes BACILLOS GRAM+ Escherichia coli Haemophilus influenzae Legionella pneumoniae phitia Proteus mirabilis Salmonella typhi Shigella spp OTROS Pneumocystis joroveci ANTISÉPTICOS/ANTIMICROBIANOS DEL TRACTO URINARIO Reacciones adversas Dermatologicos: las reacciones cutáneas Gastrointestinales: náuseas, vómitos, glosistis y estomatitis Hematológicos: anemia megaloblástica, leucocitopenia y trombocitopenia Pacientes con VIH: neumonía por P. jiroveci, fiebre, axantemas, diarrea o pancitopenia Fármaco METENAMINA Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Se descompone en un pH acido urinario, igual o inferior a 5,5, y producir formaldehido, que es toxico para la mayoría de las bacterias. La reacción es lenta para lograr una descomposición del 90% se necesitan 3h. No se debe usar en pacientes con sondas vesicales a permanencia. Las bacterias no desarrollan resistencia a esta Se administra por v.o Se distribuye por todos los líquidos corporales, pero no se descompone a un pH de 7,4 y no se produce toxicidad sistemita Se elimina por la orina Las bacterias que descomponen la urea, como Proteus spp. Por lo general son resistentes Se utiliza para tratamiento supresor crónico. Este se emplea para el tratamiento de las ITUS bajas Reacciones adversas Molestias gastrointestinales En dosis altas puede aparecer albuminuria, hematuria y exantemas NITROFURANTOINA Fármaco Nitrofurantoina Farmacodinamia Las bacterias sensibles reducen el medicamento a un intermediario altamente activo que inhibe varias enzimas Farmacocinética Se administra por V.O Resistencia A bacterias Gram- comunes en el tracto urinario Espectro antibacteriano E. colis s. saprophyticus Reacciones adversas Anemia hemolítica en pacientes con deficiencia de G6PD. Molestias digestivas Neumonitis agudas Problemas neurológicos Fibrosis pulmonar intestinal en pacientes que reciben nitrofurantoina a largo plazo No se debe administrar en pacientes con alteración renal importante o en mujeres embarazadas que tienen 38 semanas o más de embarazo ANTIMICOBACTERIANO ISONIAZIDA Fármaco Isoniazida Farmacodinamia Es un profármaco que se activa gracias a la acción de una catalasa-peroxidasa micobacteriana Farmacocinética Resistencia V.O La absorción se altera si se administra con alimentos sobre todo grasos Se difunden en todos los líquidos corporales, células y material ceroso del organismo. Las concentraciones del fármaco en el LCR son Mutaciones cromosómicas Mutaciones de la proteína transportadora de acilo Sobreexpresion de la enzima efectora INHA Resistencia cruzada entre isonizida y la etionamida Espectro antibacteriano Mycobacterium tuberculosis Mycobacterium kensasii puede ser sensible a concentraciones elevadas medicamento Reacciones adversas Hepatitis es el efecto mas grave que si no se diagnostica y la administración de la isoniazida si continua puede llegar a ser mortal. Su incidencia aproximadamente iguales a las del suero. El fármaco penetra fácilmente en las células huésped y es eficaz contra los bacilos que crecen intracelularmente El tejido infectado tiene a conservar el fármaco durante mas tiempo La isoniazida experimenta Nacetilacion e hidrolisis y se forman productos aumenta con la edad y en aquellos que reciben además rifampicina o ingieren alcohol a diario La neuropatía periférica se manifiesta como parestesias en las manos y en los pies es secundaria a una deficiencia de piridocxina (vit B6) Convulsiones en pacientes propensos a ellas. Hipersensibilidad exantema y fiebre RIFAMPICINA Fármaco Rifampicina Farmacodinamia Farmacocinética Bloquea la transcripción a través de la interacción con la subunidad B de la ARN polimerasa bacteriana dependiente de ADN, pero no con la humana. Administración: V.O Se distribuye por todos los líquidos y organos corporales. EN el LCR se alcanzan concentraciones suficientes, incluso en ausencia de inflamación Resistencia Puede producirse como consecuencia de una mutación que afecta a la afinidad de la ARN polimerasa bacteriana dependiente de ADN por el fármaco Espectro antibacteriano Reacciones adversas Es bactericida, tanto para las bacterias intracelulares como para las extracelulares, incluidas M. tuberculosis y las micobacterias atípicas, como M. kansasii Es eficaz contra microorganismos Gram+ y Nauseas Vómitos Exantemas La hepatitis y la muerte por insuficiencia hepática son raras Es captado por el hígado y sigue el ciclo enterohepático La eliminación se da a través de la bilis a las heces o por vía urinaria Gram- y se utiliza con frecuencia profilácticamente en los individuos expuestos a la meningitis producida por meningococos o Haemophilus inlfuenzae Sirve contra la lepra Cuando se administra de un modo intermitente o en dosis diarias de 1,2 g o mayores con frecuencia aparece un síndrome de tipo gripal, escalofríos y mialgias, en ocasiones se asocia con insuficiencia renal aguda, anemia hemolítica y choque RIFABUTINA Fármaco Rifabutina Farmacodinamia Derivado de la rifampicina Farmacocinética Resistencia Espectro antibacteriano Fármaco de elección en los pacientes tuberculosos infectados por VIH Reacciones adversas Similares a los de a rifampicina, pero también pueden causar uveítis, hiperpigmentació n cutánea y neutrocitopenia RIFAPENTINA Fármaco Rifapentina Farmacodinamia Farmacocinética Resistencia Actividad comparable a la de la rifampicina, su semivida es más prolongada que la de esta y la rifabutina Puede administrarse semanalmente Para la fase intensiva del tratamiento breve para la tuberculosis (2 primeros meses) Se administra 2 veces por semana EN la fase siguiente se administra una vez por semana durante 4 meses Para evitar la resistencia no se debe administrar sola, sino en una pauta de 3 o 4 fármacos Espectro antibacteriano Reacciones adversas Espectro antibacteriano Reacciones adversas PIRAZINAMIDA Fármaco Pirazinamida Farmacodinamia Es un agente sintético y se desconoce su mecanismo de acción Farmacocinética Es un antituberculoso bactericida sintético, eficaz por v.o que se emplea con combinación con la isoniazida, la rifampicina y el etambutol Se distribuye por todo el organismo y penetra el LCR Se metabolisa de forma completa Resistencia Cepas resistentes carecen de pirazinamidasa Se activa contra los bacilos tuberculosos en el ambiente acido de los lisosomas y en los macrófagos Cerca del 15% de los pacientes que reciben isoniazida, rifampicina y pirazinamida pueden presentar disfunción hepática También retención de uratos y se puede dar ataque de gota ETAMBUTOL Fármaco Etambutol Farmacodinamia Es bacteriostático Inhibe la arabinosiltransferasa, enzima para la síntesis de la pared celular micobacteriana de arabinogalactano Farmacocinética Su absorción por v.o Se distribuye por todo el organismo. La penetración en el SNC es terapéuticamente suficiente para la meningitis tuberculosa Resistencia Espectro antibacteriano No constituye un problema si se administra el fármaco junto con otros agentes antituberculosos Reacciones adversas Neuritis óptica con visión borrosa Ceguera para los colores rojo-verde FARMACOS ALTERNATIVOS DE SEGUNDA LINEA Fármaco Estreptomicina Farmacodinamia Primer antibiótico eficaz en el tratamiento de la tuberculosis Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas CAPREOMICINA Fármaco Capreomicina Farmacodinamia Antagoniza los pasos de la síntesis de la pared celular bacteriana en los que interviene la Dalanina Farmacocinética Resistencia Administración: parenteral Espectro antibacteriano Reacciones adversas Se reserva principalmente para el tratamiento de la tuberculosis multirresistente. Su administración requiere un control cuidadoso del paciente para prevenir la nefrotoxicidad y la ototoxicidad del fármaco. CICLOSERINA Fármaco Cicloserina Farmacodinamia Antagoniza los paso de a síntesis de la pared celular bacteriana en los que interviene la D-alanina Farmacocinética Eficaz por v.o Se distribuye bien por todos los líquidos corporales, incluidos el LCR Se metaboliza y se excreta junto con sus metabolitos a través de la orina Resistencia Espectro antibacteriano Agente antituberculostatico Reacciones adversas Los efectos adversos consisten en alteraciones del SNC, con posible exacerbación de las convulsiones epilépticas. También pueden producirse neuropatías periféricas, aunque ETIONAMIDA Fármaco Etionamida Farmacodinamia Analogo estructural de la isoniazida, no actúa al mismo mecanismo Inhibe la acetilación de la isoniazida Farmacocinética Resistencia Espectro antibacteriano Es eficaz por v.o y se distribuye ampliamente por el organismo, incluido el LCR Se metabolisa completamente y se elimina por la orina Reacciones adversas Irritación gástrica, hepatotoxicidad Nuropatias periféricas Nuritis óptica FLUOROQUINOLONAS Fármaco Fluoroquinolonas Farmacodinamia La moxifloxacina y la levofloxacina, ocupan un lugar importante en el tratamiento de la tuberculosis multirresistente. Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas MACRÓLIDOS Fármaco Macrólidos Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas La azitromicina y la claritromicina, forman parte de la pauta que incluye también al etambutol y la rifabutina, que se utiliza para el tratamiento de las infecciones por el complejo M. avium-intracellulare. La azitromicina es preferible en los pacientes infectados por el VIH, porque la probabilidad de que interfiera en el metabolismo de los fármacos antirretrovvíricas es menor. FARMACOS USADOS PARA TRATAR LA LEPRA DAPSONA Fármaco Farmacodinamia Farmacocinética Resistencia Espectro antibacteriano Reacciones adversas Dapsona Relacionada estructuralmente con las sulfamidas inhiben la dihidropteroato sintetasa en la via de la síntesis del folato bactericida Se administra por v.o Se absorben en el aparato digestivo Se distribuye por todo el organismo con concentraciones elevadas en la piel Experimenta acetilación hepática Se elimina por la orina M. leprae Pneumosytis jiroveci Hemolisis en pacientes con deficiencia de G6FD metahemoglobinemia y neuropatía periférica CLOFAZIMINA Fármaco Clofazimina Farmacodinamia Es un colorante de fenazina. Su mecanismo de acción implica en la unión con el ADN. Es bactericida Farmacocinética Administración: V.O Se acumula en los tejidos y esto permite un tratamiento intermitente; no penetra en el SNC Resistencia Espectro antibacteriano M. leprae M. aviarium-intracellulare Reacciones adversas Enteritis esosinofilica El fármaco también es antiinflamatorio y evita el desarrollo del eritema nudoso leproso. Referencia bibliográfica 1. Clark M, Finkel R, Rey J, Whalen K. Farmacología [Internet]. 5.ª ed. Barcelona, España: Richard A. Harvey; 2012 [citado 26 noviembre 2020]. Disponible en: https://drive.google.com/file/d/1vhCI_-JSWLGAyJ0Uzay9J9zVHipJvSoQ/view