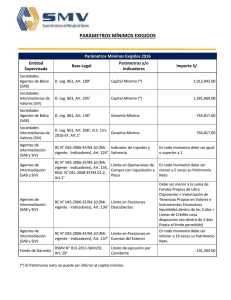
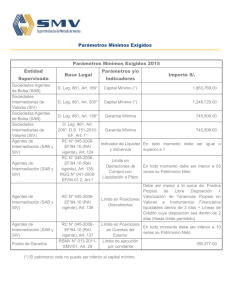
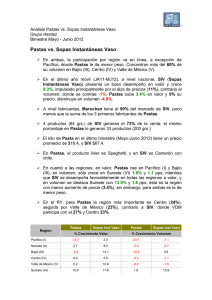
Análisis filogenético del Virus de Inmunodeficiencia en Humanos
Anuncio

Análisis filogenético del Virus de Inmunodeficiencia en Humanos (HIV) y en Simios (SIV) (Retroviridae) Castellanos, D., Chacón, A., Gómez, N., Sanabria, O. Resumen El virus HIV perece tener su origen en África central a partir de infecciones procedentes de cepas de SIV. El presente trabajo reconstruye la filogenia del virus a partir del genoma de 38 cepas HIV-1, HIV-2, SIV y FIV, realizando análisis Bayesiano, Parsimonia y Máxima Verosimilitud (ML) y. Las topologías halladas sustentan monófila del clado HIV-1, relacionado con el grupo SIVcpzptt, y del clado HIV-2 relacionado con el grupo SIVsmm. Algunas de las cepas del SIV revelan tasas significativas de cambio, mostrando una relación no cercana con las demás cepas del SIV. Introducción El HIV y SIV pertenecen a los lentivirus de la familia Retroviridae, que se caracteriza por codificar una polimerasa llamada transcriptasa inversa; el HIV es uno de los agentes patógenos con mayor tasa de variabilidad genética y con una alta capacidad de adaptación en su hospedero humano [1], esta diversidad se refleja en la existencia de diferentes grupos M, N y O [2], y hasta 32 especies recombinantes [3], cada uno de los cuales parece haber surgido de manera independiente en África central a partir de infecciones procedentes de cepas de SIV[4]. Se puede decir que el HIV es una de las entidades biológicas mejor estudiadas, lo que no implica, que todas las preguntas sobre su origen y evolución hayan sido resueltas en forma satisfactoria [5]. Este estudio pretende evaluar si SIV y HIV son grupos hermanos y las relaciones internas de los mismos utilizando datos moleculares. Metodología El análisis filogenético se realizó a partir de genomas completos de 35 cepas HIV-1, HIV2, SIV y tres taxas outgropus, las secuencias y los números de accesión se descargaron del GenBank (Tabla 1). Se aplicó un análisis de sensibilidad de las secuencia nucleotidicas con POY5 [6], para tres sets de costos diferentes (costo 1 = ts1tv1g1; costo 2 = ts1tv2g2, costo 3 = ts1tv2g4) y se determinó cual se ajustaba mejor a los datos según las tasas asignadas a transición, transversión y gaps. A continuación se seleccionó el costo más adecuado utilizando el índice de congruencia de Farris [7], posteriormente se realizó un análisis de parsimonia con TNT 1.1 [8], con un hold de 10000 y boostrap de 100. Adicionalmente se realizó el alineamiento de los datos moleculares con el programa SeaView 4 (opción Muscle) [9]; se determinó el modelo evolutivo para cada gen (gag, pol, vif, vpx, vpr, env, nef) y para el genoma completo mediante jModelTest 3.7 [10] bajo el criterio de Akaike (AIC). A continuación se realizó el análisis de Máxima Verosimilitud (ML) para el genoma, con una estrategia de búsqueda de la topología basada en algoritmos NNI y SPR, utilizando PhyML 3.0 [11]. El análisis Bayesiano fue realizado usando MrBayes 3.1.2, [12] por ocho millones de generaciones, con cuatro cadenas de Markov-Montecarlo bajo el algoritmo de Metropolis-Hasting (MCMC), esto para el genoma completo. Los árboles se visualizaron con el programa Figtree 1.2.2 [13]. Resultados y Discusión El mejor costo del análisis de sensibilidad se obtuvo con valores de ts1tv1g1 para los genes y el genoma. De acuerdo con del índice de incongruencia de Farris la interacción de costos óptimos presento un ILD=0,192 (Tabla 2). El modelo de sustitución nucleotídica obtenido con JModelTest fue GTR+G+I para cada gen y el genoma. Las estimaciones de las relaciones filogenéticas bajo el criterio de máxima verosimilitud (ML) para el genoma señalaron monofila para el grupo HIV1 y para el grupo HIV2, cada uno relacionado con SIVcpz y SIVsmm respectivamente (Figura 1). Los grupos de estudio bajo este análisis arrojan un buen soporte, con tazas evolutivas de cambio bajas: 0.339 para SIVcpz-HIV1 y 0.044 para SIVsmm-HIV2, lo cual es congruente con las teorías de evolución del virus según Sharp & Hahn [14], Charleston & Robertson [15], Wertheim & Worobey [16] y Lucie & Martine [17], evidenciando una variabilidad adecuada en las secuencias analizadas lo que permitió obtener una buena resolución en la topología de los taxones estudiados, congruente con los artículos anteriormente citados. El análisis de inferencia bayesiana para la evidencia total (Figura 2), representó una topología con un buen soporte de probabilidad posteriori en toda la filogenia; sin embargo, se presenta una politomía que disminuye el poder explicativo del cladograma, debido a que no permite dilucidar las relaciones de parentesco entre los grupos dentro de la politomía. Esto pudo deberse a un muestreo pobre a causa de la falta de poder computacional disponible para la resolución de dicha topología, por lo que no se relacionó debidamente la presencia de la variabilidad entre secuencias. La topología es el producto de un consenso de la mayoría para solucionar los conflictos entre los árboles disponibles en el programa. Sin embargo, los taxas terminales propias del grupo HIV1 relacionados con SIVcpz y el grupo HIV2 relacionados con el SIVsmm son consistentes respectivamente con los resultados obtenidos por Paraskevis et al [18] y Santiago et al [19]. El análisis de parsimonia realizado con TNT 1.1 se obtuvo un árbol consenso de longitud de 22239 pasos (Figura 3). Los valores de remuestreo de Bootstrap dan buen soporte en todos los nodos de la topología. Existe consistencia en las tres topologías halladas por parsimonia, máxima verosimilitud e inferencia bayesiana en cuanto a la monofilia del clado HIV1 el cual se encuentra relacionado con el grupo SIVcpzptt. La monofilia del clado HIV2 que está emparentado con el grupo SIVsmm está soportada en parsimonia y máxima verosimilitud. Conclusiones En la hipótesis del origen del HIV, propuesta por Sharp & Hahn [14], Charleston & Robertson [15], Wertheim & Worobey, [16], Lucie & Martine [17], Heeney et al. [22,] plantean al SIV como el origen del HIV, siendo el HIV producto de eventos de trasmisión de distintos SIV desde primates africanos no-humanos a humanos; el SIV de Pan troglodytes troglodytes dio lugar a los grupos M, N, y O del HIV1, por otra parte HIV2 surgió a partir de cepas del SIVsmm provenientes del primate no-humano Cercocebus sp [21]. Lo anterior plantea que el HIV y SIV no son grupos hermanos, esto se evidencia en las topologías halladas en los análisis de este estudio, en las cuales se encuentran taxas terminales correspondientes a algunas cepas de SIV que presentan tasas significativas de cambio, mostrando una relación no cercana con las demás cepas del SIV. Concluimos que el HIV es producto de la variabilidad, mutación y adaptación de SIV aun nuevo hospedero, el humano, esto se debe a la alta tasa mutacional presente en los genes de la cápside (gag), envoltura (env) y polimerasa inversa (pol) que causa la gran heterogeneidad del virus, pero aun así, los datos son insuficientes para proporcionar una conclusión confiable sobre ello. Recomendaciones Se recomienda el uso del programa TNT 1.1, para el análisis de parsimonia, debido a que es un programa con algoritmos de búsquedas de árboles muy rápido, con una extensiva capacidad de manejo y diagnosis del mismo [23]. Bibliografia 1. Requejo HI. 2006. Worldwide molecular epidemiology of HIV. Rev Saude Publica. 40(2): 331-45. 2. Kalish ML, Robbins KE, Pieniazek D & col. 2004. Recombinant viruses and early global VIH- epidemic. Emerging infectious diseases 10:1227-1234. 3. Kuiken C, Thakallapalli R, esklid A & de Ronde A. 2000. Genetic analysis reveals epidemiologic patterns in the spread of human immunodeficiency virus. Am J Epidemiol. 152(9): 814-22. 4. Nájera R, Delgado R, Pérez_Álvarez L & col. 2002. Genetic recombination and its role in the development of the VIH-1 pandemic. AIDS 16:S3-S16. 5. Carvajal A, Keith A. Crandal & Posada D. 2005.Caracterización y modelado computacional de la recombinación en secuencias de ADN en el virus del SIDA (VIH-1) Biojournal (2). 6. Varón A, Lucaroni, L. & Hong, W. 2011. POY 5.0.Black Sabbath Development build 6ed6dd92b5f3 R.C. New York, American Museum of Natural History. L. M. Crowley, M. Cevasco, J. S. S. Denton. 7. Farris J, Kallersjo M, Kluge, A. & Bult C. 1995. Constructing a significance test for incongruence. Syst. Biol. 44: 570-572. 8. A. Goloboff, Farris, & Nixon. 2008. Cladistics 24:774-786. 9. Gouy M, Guindon S, and Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27: 221-224. 10. Posada D. & Crandall K, 1998. Modeltest: testing the model of DNA substitution. Bioinformatics 14: 817-818 11. Guindon S. & Gascuel O., 2008. A simple, fast and accurate algorithm to estímate large phylogenies by maximun likelihood. Systematic Biology 52, 696-704. 12. Huelsenbeck J & Ronquist F. 2001. MrBayes v3.1.2: Bayesian inference of phylogeny. Bioinformatics 17: 754-755. 13. Rambaut, A. 2009. Tree Figure Drawing Tool. Versión 1.2.3. Institute of Evolutionary Biology, University of Edinburgh. 14. Sharp P. & Hahn B., 2011. Origins of HIV and the AIDS Pandemic. Cold Spring Harb Perspect Med 2011; doi: 10.1101/cshperspect.a006841 15. Charleston MA, Robertson DL. 2002.Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst Biol. 51 : 528-35. 16. Wertheim JO, Worobey M. 2007. A challenge to the ancient origin of SIVagm based on African green monkey mitochondrial genomes. PLoS Pathog. 17. Lucie E & Martine P, 2010, Origine du VIH, une réussite émergentielle, Institut de recherche pour le développement, Université de Montpellier, France, Virologie, 14 (3) : 171-8. 18. Paraskevis D., Lemey P., Salemi M., Suchard M., Van der Peer Y., & Vandamme M.2003, Analysis of the evolutionary relationships of HIV-1 and SIVcpz sequences using Bayesian inference: Implications for the origin of HIV-1. Society for Molecular Biology and Evolution. 19. Santiago M., Range F., Keele B., Li Y., Bailes E., Bibollet F., Fruteau C., Noe R., Peeters M., Brookfield J., Shaw G., Sharp P. and Hahn B. 2005. Simian Immunodeficiency Virus Infection in Free-Ranging Sooty Mangabeys (Cercocebus atys atys) from the Taı¨ Forest, Coˆte d’Ivoire: Implications for the Origin of Epidemic Human Immunodeficiency Virus Type 2. Journal of virology, Oct. p. 12515–12527, 20. Alcamí J. 2004, Avances en la inmunopatología de la infección por el VIH. Unidad de Inmunopatología del Sida. Centro Nacional de Microbiología. Instituto de Salud Carlos III. Majadahonda. Madrid. España.22(8):486-96 21.Wertheim JO, Worobey M. 2009. Dating the Age of the SIV Lineages That Gave Rise to HIV-1 and HIV-2. PLoS Comput Biol 5. 22. Heeney. J, Dalgleish. A, Weiss. R, 2006. Origins of HIV and the Evolution of Resistance to AIDS. DOI: 10.1126/science.1123016 Science 313, 462. 23. Goloboff, 1999, Cladistics 15:407-428 Anexos Tablas Código de accesión EU117991.1* Secuencia FIV EU117991.1 Pantera leo FIV EF455614 Puma concolor FIV U56928 Felis manul Abreviatura outgroupa Año 2008 Ubicación geográfica Congo, Zambia, Angola y Sudáfrica outgroupc 2008 América outgroupb 1997 HIV1 AB485633 Ghana HIV1 AY271690 Cameroon HIV1 AJ866555 Cote d'Ivoire HIV1 AY322190 Kenya HIV1 AF197341 Central African HIV1 AF069671 Uganda HIV1 AF361873 Tanzania HIV1 AJ251057 Senegal HIV1 FM877778 Republica democratica SIV HQ378594.1 Chlorocebus sabaeus HIV1Ghana 2006 Entre los 1000 y 4000 metros en Irán, India, Pakistán, Afganistán, China y Mongolia. Ghana HIV1Cameroon 2003 Camerón HIV1CotedIvoire 2005 Cote d´Ivoire HIV1Kenya 2004 Kenya HIV1Central África HIV1Uganda 2002 África Central 2000 Uganda HIV1Tanzania 2001 Tanzania HIV1Senegal 2006 Senegal HIV1 Repubdemocr 2009 República democrática SIVChlorosab a 2011 África subsahariana (desde Senegal: Sudáfrica) M66437.1 SIV_M66437.1 Chlorocebus sabaeus SIVChlorosab b 2008 África subsahariana (desde Senegal: Sudáfrica) M58410.1 SIV M58410.1 Chlorocebus sabaeus SIV AY159322.1 Mandrillus sphinx SIV Chlorosab c 1991 África subsahariana (desde Senegal: Sudáfrica) SIV Mandrillus sphi SIV Mandrileuc 2006 Oeste de África , desde Guinea Ecuatorial al Congo Camerún, Nigeria y Guinea Ecuatorial SIVPantroglo a 2003 África central, Sudán, Tanzania, Zambia, Uganda y R. del Congo. SIV Pantroglo b 2011 Gabón, Guinea y R. del Congo. SIV Pantrogl c 2005 Gabón, Guinea y R. del Congo. EF455614* U56928* AB231898 AY271690 AJ866555 AY322190 AF197341 AF069671 AF361873 AJ251057 FM877778 HQ378594.1 AY159322.1 AY159321.1 SIV AF447763.1 FR686510.1 X52154.1 SIV AY159321.1 Mandrillus leucophaeus SIV AF447763.1 Pan troglodytes schweinfurthii SIV FR686510.1 Pan troglodytes troglodytes SIV X52154.1 Pan troglodytes troglodytes 2006 SIV AF103818.1 Pan troglodytes troglodytes SIV FM165200.1 Procolobus verus SIV Pantrogl d 1999 Gabón, Guinea y R. del Congo. SIV Procoverus 2010 SIV AF301156.1 Colobus guereza SIV M19499.1 Macaca mulatta SIV Colobgue 2001 SIVMaccamul 2000 Costa de Marfil, Ghana, Guinea, Liberia, Nigeria y Sierra Leona. África oriental y central, Etiopía, Tanzania, Nigeria, Zambia y Chad. Etiopía SIV L06042.1 Cercopithecus mitis SIV U58991.1 Cercopithecus tantalus SIVCercopi a 2000 SIVCercopi b 2005 HM803689.1 SIV HM803689.1 Cercocebus torquatus SIV Cercopi c 2010 NC001549 SIV NC001549.1 Cercopithecus aethiops SIV FR751162.1 Cercopithecus solatus SIV AF075269.1 Cercopithecus l'hoesti SIV Cercopi d 2009 SIVCercopi e 2011 Selva humedad en Gabón SIV Cercopi f 1999 Uganda, Burundi, Ruanda y el Congo HIV2 M30502 Mali HIV2 X52223 Gambia HIV2 J04542 Ghana HIV2 D00835 Guinea Bissau HIV2 AF208027 Cote d`Ivoire HIV2 EU028345 Camerón HIV2 M15390 Senegal HIV2 U27200.1 Costa de marfil HIV2 Mali HIV2 Gambia 2002 2005 Mali Gambia HIV2 Ghana HIV2 GuiBiss 2000 2007 Ghana Guinea Bissau HIV2 CotdIvoi 2000 Cote d` Ivoire HIV2 Camero 2008 Camerón HIV2 Senegal 1996 Senegal HIV2 Costmarf 1995 Costa de Marfil AF103818.1 FM165200.1 SIV AF301156.1 M19499.1 L06042.1 U58991.1 FR751162.1 AF075269.1 M30502 X52223 J04542 D00835 AF208027 EU028345 M15390 U27200.1 Entre el Valle del Rift y río Congo hasta Angola y Zambia. Ghana, Sudan y Kenia. Camerún, R.D.Congo, Costa de Marfil, Guinea, Gabón , Ghana, Nigeria, Senegal y Sierra Le África subsahariana (desde Senegal: Sudáfrica) Tabla 1. Listado de cepas con sus respectivas abreviaturas y números de accesión para los genomas usados en el presente estudio. Las especies marcadas con asterisco (*) fueron consideradas como outgroup. Costos ET Env Gap Pol Vif Vpx Vpr Nef,Tat,Env ILD 111-111-1 66028 1737 9 8965 1616 2 107 2 201 0 206 9 5689 0,19207003 122-1222 10589 0 2764 1 1387 5 2457 9 175 3 329 2 335 5 9199 0,20961375 124-1244 13289 4 3320 2 1644 7 2766 6 217 0 421 5 407 8 11486 0,25305883 Tabla 2. Índice de incongruencia de Farris (ILD) para con los tres sets de costos asignados a los Genes Env, Gap, Pol, Vif, Vpx, Vpr respectivamente. Se resalta el valor de ILD óptimo. Ilustraciones Figura 3. Filogenia de lentivirus. Mostrando las relaciones evolutivas en las secuencias genómicas de HIV y SIV derivados de varios huéspedes primates; El grupo externos son miembros de la familia felidae y están con los siguientes nombres: “Outpantera”, “Outfelis” y “Outpuma”. Los virus de la cepa SIVcpz están señalados con azul, los virus de la cepa SIVsmm con rojo. Este árbol fue estimado usando el método de parsimonia. Figura 2. Filogenia de lentivirus. Mostrando las relaciones evolutivas en las secuencias genómicas de HIV y SIV derivados de varios huéspedes primates; El grupo externo son miembros de la familia felidae y están con los siguientes nombres: “Outpantera”, “Outfelis” y “Outpuma”. Los virus de la cepa SIVcpz están señalados con azul, los virus de la cepa SIVsmm con rojo. Este árbol fue estimado usando el método de inferencia bayesiana. Figura 1. Filogenia de lentivirus. Mostrando las relaciones evolutivas en las secuencias genómicas de HIV y SIV derivados de varios huéspedes primates; El grupo externo son miembros de la familia felidae y están con los siguientes nombres: “Outpantera”, “Outfelis” y “Outpuma”. Los virus de la cepa SIVcpz están señalados con azul, los virus de la cepa SIVsmm con rojo. Este árbol fue estimado usando el método de máxima verosimilitud (ML).