Introducción
Anuncio

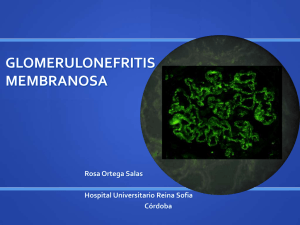
Glomerulonefritis_membranosa GLOMERULONEFRITIS MEMBRANOSA Contenido • 1 Introducción • 2 Patogenia • 3 Anatomía Patológica • 4 Características clínicas • 5 Bibliografía Introducción Se caracteriza por engrosamiento de las pared capilar glomerular, que es similar a la glomerulonefritis membranosa idiopática. Esta lesión se ve en el 10 al 15 % de los pacientes con nefritis lúpica, habitualmente se acompaña de proteinuria grave o síndrome nefrótico y puede aparecer simultáneamente con nefritis lúpica focal o difusa. Los depósitos granulares de anticuerpos y de complemento se pueden detectar mediante inmunofluorescencia. La microscopía electrónica muestra depósitos electrodensos que representan inmunocomplejos en las localizaciones mesangial, intramembranosa, subepitelial, o subendotelial. Todas las clases muestran cantidades variables de depósitos mesangiales. En las nefritis lúpica membranosa, los depósitos son predominantemente subepiteliales (entre la membrana basal y las células epiteliales viscerales). Se ven depósitos subendoteliales (entre el endotelio y la membrana basal) en los tipos proliferativos (clases III y IV), aunque se pueden encontrar con poca frecuencia en las clases I, II y V de las nefritis lúpica. Cuando son prominentes, los depósitos subendoteliales crean un engrosamiento difuso de la pared capilar, que se ve en microscopia óptica como una lesión en asa de alambre. La nefropatía membranosa es una causa frecuente de síndrome nefrótico en adultos. Se caracteriza por el engrosamiento difuso de la pared capilar glomerular debido a la acumulación de depósitos electrodensos de inmunoglubolina siguiendo la vertiente subepitelial de la membrana basal. La glomerulopatía membranosa que se presenta asociada a otras enfermedades sistémicas y a varios agentes etiológicos identificables se considera una glomerulopatía membranosa secundaria. Las asociaciones más notables son las siguientes: • Fármacos ( penicilina, captopril, oro, fármacos antiinflamatorios no esteroideos (AINE): el 1- 7% de los pacientes con artritis reumatoide trtados con penicilina u oro (fármacos que ahora no son muy utilizados en esta indicación) desarrollan una glomerulopatía membranosa. Los AINE, como veremos más adelante tambiñen causan enfermedad de cambios mínimos. • Tumores Malignos subyacentes, en particular carcinomas de pulmón, colon y melanoma. De acuerdo con algunos investigadores, se presentan hasta en el 5-10% de los adultos con una glomerulopatía membranosa. • LES. Aproximadamente el 10-15% de las glomerulonefritis del LES son del tipo membranoso. • Infecciones (hepatitis B crónica, hepatitis C, sífilis, esquistosomiasis, malaria). • Otros trastornos autoinmunitarios, como la tiroiditis, pueden detectarse en una glomerulopatía membranosa secundaria. En aproximadamente el 85% de los pacientes no se puede detectar ningún trastorno asociado y se consideran idiopáticos. Introducción 1 Glomerulonefritis_membranosa Patogenia La glomerulopatía membranosa es una forma de enfermedad crónica mediada por complejos inmunitarios. En la glomerulopatía membranosa secundaria los antígenos desencadenantes a veces se pueden identificar en los complejos inmunitarios. Por ejemplo, la glomerulopatía membranosa del LES se asocia al depósito de complejos autoantígeno-anticuerpo. Los antígenos identificados en los depósitos de algunos pacientes son los antígenos exógenos, antígenos endógenos no renales y antígenos endógenos renales. Las lesiones son notablemente semejantes a las de la nefritis experimental de Heymann que se induce por anticuerpos frente a un complejo antígeno megalina. Aún no se sabe si existe un antígeno similar en la mayoría de los casos de glomerulopatía membranosa idiopática en el hombre. La susceptibilidad de la nefritis de Heymann en ratas y la glomerulopatía membranosa en el hombre está relacionada con el locus del complejo mayor de histocompatibilidad, que puede influir en la capacidad de producir anticuerpos frente al antígeno nefritógeno. Por tanto, la glomerulopatía membanosa idiopática, como la nefritis de Heymann, se considera una enfermedad autoinmunitaria relacionada con los genes de susceptibilidad y se debe principalmente a anticuerpos frente a un autoantígeno renal. Las fugas por la pared capilar glomerular en la glomerulopatía membranosa se produce a partir del complemento. Los neutrófilos, monocitos o plaquetas son escasos en los glomérulos. La presencia practicamente uniforme de complemento y los estudios experimentales indican la acción directa de del componente C5b-C9, la vía que conduce a la formación del complejo de unión a la membrana. Se ha propuesto que ese complejo C5b-C9 activa las células epiteliales glomerulares y mesangiales, induciendo la liberación de proteasas y oxidantes que causarían la lesión de la pared capilar y el aumento de la pérdida de proteínas. Anatomía Patológica Con el microscopio óptico, los glomérulos tienen un aspecto normal en los primeros estadios de la enfermedad o muestran un engrosamiento difuso de la pared capilar glomerular. Con el microscopio electrónico se comprueba que el engrosamiento se debe a depósitos irregulares de complejos inmunitarios entre la membrana basal y las células epiteliales supraadyacentes, estas últimas con afectación a los podocitos. El material de la membrana basal se localiza entre esos depósitos, con el aspecto de espículas irregulares que hacen protusión desde la MBG. Estas espículas se ven mejor con las tinciones de plata, que tiñen la membrana basal pero no los depósitos, negros. Con el tiempo, esas espículas se engruesan para producir protusiones a modo de cúpulas y, finalmente, se cierran sobre los depósitos inmunitarios, enterrándolos dentro de una membrana irregular y muy engrosada. Con el microscopio de inmunofluorescencia, se demuestra que los depósitos granulares contienen tanto inmunoglobulinas como complemento. A medida que avanza la enfermedad se puede producir esclerosis. A lo largo del tiempo, los glomérulos pueden esclerosarse por completo. Las células epiteliales de los túbulos proximales contienen gotículas por la reabsorción de proteínas y puede verse una considerable inflamación intersticial con células mononucleadas. Características clínicas En un individuo previamente sano, el síndrome nefrótico en este trastorno tiene un inicio insidioso o, en el 15 % de los pacientes, con proteinuria no nefrótica. La hematuria y la hipertensión leve se presentan en el 15-35% de los casos. Es necesario descartar siempre las causas secundarias, ya que el tratamiento de la afección subyacente (neoplasia maligna, infección o LES) o la retirada del fármaco agresor pueden revertir la lesión. La evolución de la enfermedad es variable, pero generalmente indolente. La proteinuria no es muy selectiva, y no responde bien a los corticoesteroides. La progresión se asocia a un aumento de la esclerosis de los glomérulos y el incremento de Características clínicas 2 Glomerulonefritis_membranosa la creatinina sérica refleja la insuficiencia renal y el desarrollo de la hipertensión. Aunque la proteinuria persiste en más del 60% de los pacientes, sólo el 10 por ciento fallecen o progresan a la insuficiencia renal antes de 10 años y no más del 40% desarrollarán finalmente la insuficiencia renal. La esclerosis concurrente de los glomérulos en la biopsia renal en el momento del diagnóstico es un factor predictivo del mal pronóstico. Las remisiones espontáneas y la evolución relativamente benigna son más frecuentes en mujeres y en sujetos con proteinuria en el rango no nefrótico. Dada la evolución variable de la enfermedad, ha sido difícil evaluar la eficacia global de los corticoesteroides o de otro tratamiento inmunosupresor en el control o progresión de la proteinuria. Bibliografía 1. Kumar, V.; Abbas, A.K.; Fausto, N.; Aster, J. (2010). Robbins y Cotran. Patología Estructural y Funcional. Octava Edición. Barcelona: Elsevier. Bibliografía 3