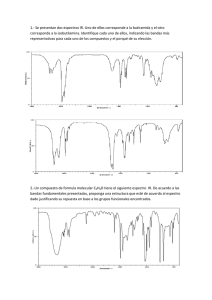
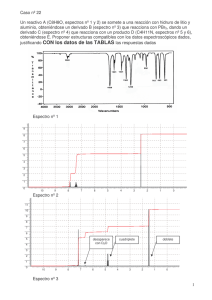
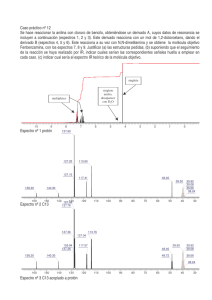
1.1 Espectroscopía de Infrarrojo
Anuncio

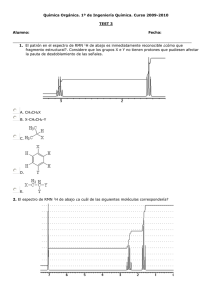
UNIDAD TEMÁTICA 1 TÉCNICAS ESPECTROSCÓPICAS 1.1 Espectroscopía de Infrarrojo Este tipo de espectroscopia se basa en la absorción de la radiación infrarroja por las moléculas en vibración. En una molécula, todos los átomos vibran alrededor de la distancia interatómica media. Existen dos modos principales de vibración, alargamiento y flexión, y éstos están cuantizados. La absorción de luz infrarroja de energía o frecuencia apropiada (2’5-15 m; 4.000-666 cm-1) excita a la molécula desde su estado fundamental hasta un estado excitado produciéndose la vibración de un modo determinado. Una molécula absorberá energía cuando ésta sea igual a la necesaria para que se produzca una transición vibracional de la molécula. Es decir, la molécula vibrará de un modo determinado gracias a la energía que se le ha suministrado. La frecuencia o longitud de onda de cada modo de absorción es función de la masa relativa de los átomos, la constante de fuerza de los enlaces y la geometría de la vibración. Esto hace posible asignar frecuencias características de alargamiento y flexión a grupos funcionales específicos, ya que, aunque las frecuencias vibracionales para un enlace dado en una molécula compleja no son totalmente independientes de los demás enlaces situados cerca, el rango de variación es pequeño. Dicho esto conviene destacar que sólo se observará un pico en el espectro de infrarrojo en el caso de que el movimiento de vibración, alargamiento o flexión, vaya acompañado de un cambio en el momento dipolar. Así mismo, cuanto más polar sea un enlace más intenso será el pico correspondiente a su frecuencia de vibración. La aplicación más habitual de la espectroscopia de infrarrojo en química orgánica es de tipo cualitativo y reside la identificación de determinados grupos funcionales de una molécula para los que se observan bandas características en determinadas regiones del espectro (véase tabla adjunta). Este hecho permite además la utilización de esta técnica en el seguimiento de una reacción en la que se tiene lugar una transformación de grupos funcionales observables en IR. En la zona del espectro con longitudes de onda comprendidas entre 1300-400 cm-1, la asignación de bandas de absorción a determinadas vibraciones moleculares es muy difícil de realizar. Esta zona es la denominada huella dactilar, característica de cada compuesto, en la que pequeñas diferencias en la estructura de la molécula dan lugar a variaciones muy importantes en los máximos de absorción. Cuestionario previo a la realización de la práctica 1 ¿Qué información nos proporciona el espectro de IR de un compuesto? 2 ¿En qué unidades se expresan las bandas de un espectro de IR? 3 ¿De qué material es la célula que se utiliza? ¿Por qué? 4 Describa la preparación de una muestra sólida para realizar un espectro de IR. 5 Busque en la bibliografía las bandas de absorción características de los grupos funcionales más representativos (alcoholes, aminas, aldehídos, cetonas, ácidos carboxílicos...) Sección experimental Reactivos y disolventes Bromuro potásico, tetracloruro de carbono, Nujol, cloroformo sin alcohol, ácido benzoico, anilina, alcanfor, acetanilida, difenilamina, n-butanol, benzoato de etilo. Material Espectrómetro de IR, un mortero de ágata, un par de placas de NaCl, un soporte para placas, un par de células para muestras líquidas, una prensa, pipetas, un desecador. Las muestras se pueden analizar tanto en fase gaseosa como en fase sólida o líquida. Pudiendo en este último caso tratarse de un líquido puro o una disolución. En general, la muestra se soporta entre dos placas de un material transparente a la radiación infrarroja (cloruro sódico o bromuro potásico) que posteriormente se sitúan directamente en la trayectoria del haz de luz. a) Gaseosa: Aunque no es muy habitual se puede realizar haciendo uso del material adecuado. Así, el vapor se introduce en una célula especial de 10 cm de longitud. Esta célula es de cloruro sódico, transparente a la radiación infrarroja en la región de 4000 a 667 cm-1. b) Líquida: En este caso se sitúa una gota del líquido entre dos placas de cloruro sódico. c) Disolución: La muestra se disuelve en tetracloruro de carbono o en cloroformo libre de alcohol (1-5%). Esta disolución se introduce en una célula especial también de cloruro sódico de 0’1 a 1 mm de espesor. d) Sólida: Existen dos técnicas dependiendo de la polaridad del compuesto a analizar. En el caso de compuestos apolares se suele preparar una emulsión del sólido en nujol (hidrocarburo) que se sitúa entre las dos placas de cloruro sódico. Esta emulsión se realiza mezclando, en un mortero de ágata, una gota de nujol con 1 mg aproximadamente del sólido problema previamente molido. Si la polaridad del compuesto es alta, se prepara una pastilla de bromuro potásico. Así, el sólido se muele con 50 a 100 veces su masa de bromuro potásico anhidro y la mezcla se prensa a vacío. Tras este proceso se forma de un pequeño disco transparente que se coloca directamente en el portamuestras. Éste método evita el uso del nujol para el que también se observan bandas en el espectro. Por último hemos de decir que los espectros de un compuesto medido en fase sólida y en disolución no tienen por qué ser idénticos debido a la presencia de las interacciones intermoleculares, especialmente aquellos grupos funcionales que pueden interaccionar mediante puente de hidrógeno. En cuanto a la conservación de las células de cloruro sódico, éstas son especialmente frágiles a los golpes y sensibles a la humedad por lo que deben limpiarse con disolventes anhidros y cuando no se utilicen guardarse en un desecador. Por otro lado, el bromuro potásico debe usarse siempre anhidro por lo que se mantendrá en un horno o estufa. Los espectros de IR de los compuestos problema se registrarán siguiendo el procedimiento adecuado según su estado y se interpretarán los datos obtenidos. 1.2 Espectroscopía de Resonancia Magnética Nuclear La espectroscopia RMN se ha convertido en los últimos años en la técnica analítica más útil en Química Orgánica. El fenómeno de la resonancia magnética nuclear se basa en el hecho de que ciertos núcleos giran alrededor de su eje, es decir, poseen espín nuclear (I). El movimiento de giro de estas especies cargadas, provoca que lleven asociados un campo magnético débil (momento magnético), pudiendo considerarse como pequeños imanes. El espín nuclear puede tener valores enteros o fracción de dos. Así, núcleos como 1H, 19F y 13C tienen valores de espín nuclear ½, el 14N tiene 1, el 11B tiene 3/2 y el 73Ge tiene 9/2. Sin embargo, otros núcleos como 12C y 16O tienen un valor cero de espín nuclear y, por tanto, son inactivos en esta técnica. En Química Orgánica los más estudiados son el 1H y el 13C. Si un núcleo con espín nuclear 1/2 se sitúa en un campo magnético externo puede adoptar dos orientaciones diferentes, paralela o antiparalela respecto al campo aplicado, siendo la primera de menor energía. La presencia exclusiva de estas dos orientaciones se debe a que los niveles de energía implicados están cuantizados. De forma similar a la espectroscopia de infrarrojo, los núcleos pueden excitarse al nivel de energía superior suministrando la radiación electromagnética adecuada e inducir así la resonancia, fenómeno que supone simplemente invertir su orientación. Cada isótopo activo en RMN absorbe radiación de frecuencia diferente en un campo magnético constante. Este hecho hace que podamos estudiar cada uno de ellos de forma independiente. Además, la diferencia de energía entre estos dos niveles es principalmente, función del núcleo que se está irradiando y del campo magnético aplicado, siendo ésta mayor cuanto más intenso es el campo magnético externo. De lo expuesto hasta ahora podría deducirse que todos los núcleos idénticos presentes en una molécula, por ejemplo todos los protones o todos los átomos de carbono 13, absorben energía a la misma frecuencia. Sin embargo, este valor difiere ligeramente dependiendo del entorno de cada núcleo en el seno de la molécula. La presencia de las nubes electrónicas que rodean a los núcleos hacen que tras la aplicación de un campo magnético externo se cree un campo magnético pequeño local que se opone al campo aplicado, por lo que el campo real o efectivo que experimenta un núcleo es un poco menor: Hefectivo = Haplicado-Hlocal Se dice entonces que el núcleo está apantallado por la nube de electrones. Un entorno químico diferente dará lugar a una densidad y distribución electrónica distinta y, por tanto, a un campo magnético efectivo distinto lo que se traducirá en señales bien diferenciadas en el espectro para cada tipo de protón o por cada tipo de carbono dentro de una molécula. La posición exacta en el espectro de RMN a la que un determinado núcleo absorbe se llama desplazamiento químico y se mide en partes por millón (ppm). Este valor es independiente de la intensidad del campo magnético aplicado. Tanto en espectroscopia de RMN de protón como de carbono los valores de desplazamiento químico están referidos al del tetrametilsilano (TMS) al que se le ha asignado, arbitrariamente, un valor cero. Además del desplazamiento químico de las señales otras características importantes de los espectros de RMN, en especial de los de protón, que nos suministran una información muy valiosa son: 1) La intensidad de las señales (valor de integración). 2) El desdoblamiento de las señales (grado de acoplamiento). a) Resonancia magnética nuclear de protón Valores de desplazamiento químico en RMN de protón Habitualmente, en un espectro de RMN de protón los desplazamientos químicos aparecen en el intervalo 0-10 ppm. Su valor exacto, como se ha comentado anteriormente, depende del entorno químico de cada núcleo. Los protones con un mismo entorno químico se dice que son químicamente equivalentes y resuenan al mismo desplazamiento químico por lo que dan lugar a una sola señal en el espectro. La equivalencia química de los protones de un determinado compuesto está relacionada estrechamente con su grado de simetría. Los protones equivalentes deben ser intercambiables entre sí mediante una operación de simetría (plano de simetría, centro de simetría o rotación). La equivalencia o inequivalencia de los protones de una molécula se puede determinar reemplazando de forma secuencial cada uno de los átomos de hidrógeno a estudiar por un sustituyente Z. Si se obtiene el mismo producto independientemente de que protón se haya sustituido éstos son químicamente equivalentes. Integración en RMN de protón El valor del área (integración) bajo una determinada señal de un espectro de RMN de protón es siempre proporcional al número de protones responsables de dicha resonancia. Por ello, el cálculo de las integrales de todas las señales del espectro nos proporciona una información muy valiosa: el número relativo de cada tipo protones. Hay que tener en cuenta, no obstante, un error del 10% en dicho valor. Acoplamiento espín-espín en RMN de protón En términos generales el fenómeno del acoplamiento espín-espín se produce por la interacción entre núcleos no equivalentes a través del enlace químico que los conecta. Así, un núcleo es capaz de sentir las orientaciones posibles de los núcleos vecinos. El acoplamiento entre espines nucleares produce un desdoblamiento de la señal que toma la apariencia de multiplete (doblete, triplete, cuadruplete,…). La diferencia de frecuencia entre los picos individuales de un multiplete se llama constante de acoplamiento (J) y su valor absoluto se encuentra en el intervalo 0-18 Hz. La constante de acoplamiento es independiente del campo magnético externo. Para predecir el tipo de acoplamiento entre los protones de una molécula podemos hacer uso de las siguientes reglas: • • • Los protones químicamente equivalentes no se acoplan entre sí (los protones equivalentes pueden estar situados en el mismo carbono o en carbonos distintos). El grado de desdoblamiento o multiplicidad de la señal dependerá del número de protones adyacentes y se puede calcular por la regla (n+1): un determinado tipo de protón que tiene (n) protones adyacentes equivalentes presentará una señal con (n+1) picos. Esta regla es sólo valida en espectros de primer orden. Dos grupos de protones acoplados entre sí siempre tienen la misma constante de acoplamiento J. Esta información permite obtener información acerca de la conectividad entre determinados fragmentos moleculares. En algunos casos pueden observarse multipletes relativamente complejos debido a las resonancias de protones acoplados a dos o más tipos de núcleos no equivalentes. b) Resonancia magnética nuclear de carbono Aunque se trata de la misma técnica, las características de los espectros de RMN de 13C difieren notablemente de las de los espectros de protón. Esto es debido principalmente a que el carbono 13 se encuentra en muy baja abundancia (1’1%) lo que provoca una gran disminución de la sensibilidad. Los espectrómetros con transformación de Fourier han permitido que la detección de la señal de carbono 13 sea factible en un tiempo razonable pero aún así, es necesaria una cantidad de muestra diez veces mayor que la utilizada para medir un espectro de RMN de protón. Debido a esta baja abundancia, la probabilidad de encontrar dos átomos de carbono 13 situados en posiciones adyacentes es prácticamente cero, por lo que no se observa acoplamiento homonuclear (13C-13C). Sí es posible observar, no obstante, acoplamiento heteronuclear (13C-1H) aunque habitualmente éste se elimina (desacoplamiento de banda ancha de protón) pues provoca una pérdida considerable de sensibilidad así como un aumento notable de la complejidad del espectro. Gracias a este desacoplamiento, las señales observadas en los espectros de carbono son líneas estrechas de las que se determina fácilmente el desplazamiento químico. Teniendo en cuenta además que el intervalo de desplazamiento químico en un espectro de carbono es mucho mayor que en uno de protón (0-200 ppm), los espectros de carbono están en la mayoría de los casos bien resueltos y se puede asignar un singlete a cada carbono no equivalente de la molécula, incluso tratándose de estructuras complejas. Otra diferencia a tener en cuenta respecto a la RMN de protón es que, por cuestiones técnicas, las intensidades de las señales obtenidas en las condiciones habituales de medida no son proporcionales al número de átomos de carbono que contribuyen a las mismas. Este hecho dificulta la identificación de grupos de núcleos equivalentes. Para la asignación de las señales en un espectro de carbono es útil la siguiente consideración: en líneas generales, las resonancias que aparecen en un espectro de carbono son aproximadamente 20 veces mayores que las de los protones que se encuentran en el mismo entorno. Así, por ejemplo, la región en la que resuenan los protones unidos a un doble enlace carbono-carbono está sobre 5 ppm, mientras que estos átomos de carbono lo hacen sobre 100 ppm. En determinación estructural la utilidad de la RMN de carbono es sumamente importante. Aunque las resonancias de los carbonos cuaternarios son más débiles normalmente que las de los carbonos que tienen protones, existen distintas técnicas monodimensionales que permiten identificar las resonancias en un espectro de RMN de carbono de acuerdo al número de protones unido a cada carbono como los experimentos con desacoplamiento off-resonance, y los experimentos DEPT o APT. El espectro de carbono 13 off-resonance es una técnica tradicional que consiste en el desacoplamiento parcial de protones. Esto se traduce en que prácticamente sólo se observen los acoplamientos a un enlace por lo que las señales aparecen entonces como singletes, dobletes, tripletes o cuadrupletes según se deban a las resonancias de carbonos cuaternarios, metino, metileno o metilo, respectivamente. La sensibilidad de estos experimentos es bastante baja debido al desdoblamiento y ensanchamiento de las líneas. Además, en moléculas complejas, la identificación de las multiplicidades puede no ser sencilla debido al solapamiento de las mismas. La técnica DEPT, utilizada de forma rutinaria actualmente, permite obtener subespectros que contienen exclusivamente resonancias debidas a carbonos metino, metileno o metilo. En esta técnica los carbonos cuaternarios desaparecen del espectro por lo que se pueden identificar fácilmente por comparación con el espectro original. El experimento APT da lugar a espectros en los que los carbonos cuaternarios y los metilenos aparecen en la parte negativa del espectro (señales hacia abajo), mientras que los metino y metilo aparecen en la parte positiva del espectro (señales hacia arriba). Actualmente, gracias a continuos avances en este campo, se han puesto a punto técnicas bidimensionales que permiten obtener información extremadamente detallada de la estructura de la molécula. Resumen de los intervalos de los desplazamientos químicos de protón en diversas clases de compuestos (δ en partes por millón [ppm] respecto del TMS) Resumen de los intervalos de los desplazamientos químicos de carbono en diversas clases de compuestos (δ en partes por millón [ppm] respecto del TMS) Cuestionario previo a la realización de la práctica 1 ¿Qué información nos proporciona el espectro de RMN de protón de un compuesto? 2 ¿En qué unidades se expresan las señales de un espectro de RMN? 3 ¿Todos los átomos de protón y carbono 13 de una molécula absorben energía a la misma frecuencia? ¿Por qué? 4 ¿Qué característica deben cumplir necesariamente los disolventes empleados en RMN? 5 Los datos espectroscópicos del espectro de IR y RMN de protón del acetato de etilo muestran la siguiente información: IR, 1732 cm-1; 1H-RMN: 1.26 (t, 3H, J = 7.1 Hz), 2.05 (s, 3H), 4.12 (q, 2H, J = 7.1 Hz). Asigne e identifique las señales. Sección experimental Reactivos y disolventes Cloroformo deuterado, tetracloruro de carbono, sulfato sódico anhidro. Material Viales, pipetas, tubos de RMN, algodón. Los espectros de RMN se miden habitualmente en disolución por lo que un punto de vital importancia es la selección del disolvente, que debe ser deuterado. En un disolvente de estas características todos sus hidrógenos se han reemplazado por deuterio (inactivo en las condiciones en las que se mide el espectro) para que las señales de los protones del disolvente no enmascaren las de la muestra a analizar, que se encuentra en mucha menor proporción. De todos los disolventes orgánicos deuterados (acetona-d6, acetonitrilo-d3, benceno-d6, cloroformo-d, diclorometano-d2, dimetilsulfóxido-d6, metanol-d4, piridina-d5, tetrahidrofurano-d8, tolueno-d8), el cloroformo-d (CDCl3) es el más empleado. También es importante el grado de concentración de la muestra así como la eliminación de partículas sólidas en suspensión que pueden distorsionar el campo magnético estático y disminuir el grado de resolución. Las muestras de compuestos orgánicos se miden, habitualmente, en tubos de vidrio de 5 mm de diámetro externo, con una longitud de 15-25 cm. El volumen de disolución que se introduce en el tubo suele ser de 0’4-0’7 ml (véase figura). Para medir el espectro de RMN de protón de una muestra se disuelve, en un pequeño vial, 30-50 mg de compuesto en, aproximadamente, 0’5 ml de disolvente deuterado. Para eliminar partículas sólidas, esta disolución se filtra directamente al tubo de RMN haciendo uso de una pipeta Pasteur obturada con un trozo pequeño de algodón (véase figura adjunta). Si la muestra está muy diluida porque disponemos de poca cantidad de muestra, posibles residuos de agua procedentes del disolvente deuterado pueden enmascarar alguna de las señales del compuesto problema. Si este es el caso conviene añadir un poco de agente desecante (sulfato magnésico o sulfato sódico) sobre el algodón utilizado para realizar el proceso de filtración. Por último, tras situar la muestra en el tubo de RMN éste se cierra con un tapón de plástico. Cuestionario sobre la práctica realizada 1 A la vista de los espectros facilitados dibuje las estructuras de los compuestos indicando los datos espectroscópicos más significativos. 1.3 Espectrometría de Masas La espectrometría de masas es, junto con la espectroscopia de infrarrojo y la de resonancia magnética nuclear, la técnica más utilizada para la elucidación estructural de compuestos orgánicos. Los espectros obtenidos mediante esta técnica además de la masa de la molécula muestran la de los fragmentos resultantes de la ruptura de la molécula mediante un proceso de ionización. Este espectro constituye una identificación prácticamente inequívoca del compuesto analizado. Sin embargo, existe una diferencia sustancial entre esta técnica y los métodos espectroscópicos. La espectroscopia se trata de un método de análisis no destructivo ya que se basa en la excitación de las moléculas desde su estado fundamental a un estado excitado mediante radiación electromagnética de frecuencia adecuada, tras la cual la molécula vuelve al estado fundamental. Por el contrario, en un espectrómetro de masas las moléculas se fragmentan tras un proceso de ionización, por lo que la muestra no es recuperable. Aunque en la actualidad existen muchos otros métodos (FAB, ionización química, técnica electrospray, MALDI), la ionización se realiza normalmente mediante impacto electrónico, en la que se bombardea a la molécula con electrones a un valor de energía de 70 eV. Este bombardeo es capaz de provocar la emisión de un electrón de la molécula, y así ionizarla dando lugar a un ion molecular que se representa como M+●: M → M+● + e Para la formación del ion molecular es necesario alcanzar el potencial de ionización de la molécula que viene determinado por la naturaleza del orbital ocupado de más alta energía del cual sale el electrón. El orden de facilidad de abstracción de un electrón es: electrones de un par no enlazante de un heteroátomo, electrones π y electrones sigma. Los iones moleculares se pueden descomponer o fragmentar debido a un exceso de energía. El tipo y la proporción relativa de cada uno de estos fragmentos son característicos de la molécula analizada y de las condiciones del proceso de ionización, y se denomina patrón de fragmentación. Tras el proceso de ionización, se aceleran todos iones obtenidos mediante campos eléctricos que les suministran la misma cantidad de energía cinética a todos ellos. La velocidad adquirida por cada ion dependerá de su masa. Los iones pasan al analizador donde son desviados mediante campos eléctricos o magnéticos. Su desviación también será función de su masa. Variando el valor del campo aplicado podemos ir dirigiendo de forma consecutiva los iones en orden de masa creciente o decreciente hasta el colector. La detección consecutiva de los iones formados da lugar al espectro de masas. El espectrómetro de masas mide la razón masa a carga (m/z) para cada ion, y, siempre que la carga sea 1, el valor de masa al que aparece el ion molecular, o cualquier otro fragmento, coincidirá con su masa real. Sin embargo, hay que tener en cuenta que pueden existir fragmentos con carga superior a 1. Cuando un catión radical M+● se fragmenta puede evolucionar de dos formas alternativas: a) Formar un catión con un número par de electrones (A+) y un fragmento neutro con un número impar de electrones (N●): M+●→ A++ N● b) Formar un nuevo ion también con un número impar de electrones (B+●) y una molécula neutra con un número par (N): M+● → B+● + N Los procesos de fragmentación están determinados por cuatro factores principales: • • • • La fuerza de los enlaces que se escinden. La estabilidad de los fragmentos iónicos o neutros que se forman. La energía interna de los iones fragmento que van a descomponerse. El intervalo de tiempo entre la formación de un ion y su detección. Ión molecular El ion molecular se puede detectar en el espectro de masas cuando éste no posee suficiente energía para fragmentarse. Aunque permite conocer el peso molecular de la muestra hay que tener en cuenta que en el espectro pueden ser visibles varios picos asignables al ion molecular si existe un segundo isótopo de masa superior en más de una unidad a la especie isotópica más abundante. En general, M+● se refiere al ion obtenido al arrancar un electrón de la molécula compuesta por los isótopos más abundantes de los elementos que forman parte del compuesto. Es importante no confundir este valor con el ion molecular más abundante (el de intensidad mayor). Probablemente la información más valiosa de un espectro de masas además de la masa sea la composición elemental del ion molecular. Los pesos atómicos monoisotópicos no son números enteros exactos en relación al carbono, por lo que una medición de la masa lo suficientemente exacta puede definir directamente la composición elemental. Con aparatos de alta resolución es posible determinar incluso masas con 4 cifras decimales. A veces a partir de espectros de masas de baja resolución también es posible determinar la fórmula molecular o, al menos, reducir el número de fórmulas posibles, considerando las intensidades relativas de los diferentes picos isotópicos. Para determinar la fórmula molecular se calcula la intensidad del ion molecular M, expresando las de los picos M+1 y M+2 como porcentajes del primero. Estos valores se comparan con las intensidades calculadas teóricamente, tabuladas por Beynon, y descritas en la bibliografía. El método que acabamos de describir para calcular la formula molecular es aplicable sólo a compuestos de peso molecular medio y de ion molecular relativamente intenso en los que las intensidades de los picos debidos a isótopos se pueden determinar de forma fiable. Las condiciones en las que se realiza el espectro influyen en la intensidad del pico correspondiente al ion molecular. Si el espectro se ha obtenido por la técnica de impacto electrónico a un voltaje de 70 eV y el ion molecular es poco intenso se puede repetir la medida con un voltaje menor. De este modo se aumenta la proporción de iones M+● de baja energía interna, disminuyendo su facilidad de fragmentación. Además de las condiciones de medida, la intensidad del pico molecular también depende de la estabilización de la carga positiva por la molécula. Biemann estableció el siguiente orden de estabilidad decreciente: compuestos aromáticos, olefinas conjugadas, compuestos alicíclicos, sulfuros, hidrocarburos lineales, mercaptanos o tioles, cetonas, aminas, ésteres, éteres, ácidos carboxílicos, hidrocarburos ramificados y alcoholes. Otro pico importante en un espectro de masas es el (M+1)+. Éste se forma por abstracción, por el ion molecular, de un átomo de hidrógeno de las moléculas neutras no ionizadas en la cámara de ionización. Para determinados compuestos como éteres, ésteres, aminas, aminoésteres y nitrilos el ion M+1 es más estable que el ion molecular. Por último, conviene saber que si la masa molecular de una molécula neutra es impar contiene necesariamente un número impar de nitrógenos y si es par no contiene átomos de nitrógeno o bien posee un número par de ellos (regla del nitrógeno). Fragmentación La determinación de la estructura de una molécula por la mayoría de las técnicas incluye la postulación de una estructura concreta, predicción de su espectro y, por último, comparación de éste con el espectro de la molécula desconocida. En espectrometría de masas se suele proceder analizando el ion molecular y las masas de los diversos fragmentos obtenidos e intentando construir moléculas hipotéticas cuyo patrón fragmentación se ajuste al observado en el espectro. Los tipos de fragmentaciones más importantes en espectrometría de masas son: a) Ruptura de enlaces carbono-carbono. La facilidad de ruptura y, por tanto, la intensidad del fragmento resultante están directamente relacionadas con la estabilidad del carbocatión que se forma. Orden de estabilidad: CH3 < RCH2 < R2CH < R3C. b) Ruptura de enlaces carbono-heteroátomo. La introducción de un heteroátomo con electrones no enlazantes en sus órbitas exteriores, como nitrógeno oxígeno o azufre, casi siempre provoca cambios muy significativos en el espectro. Habitualmente, el sustituyente más pesado es el que se pierde en primer lugar. c) Fragmentaciones concertadas. Las proporciones relativas de los fragmentos que se forman son función directa de las estabilidades relativas de los iones, radicales y moléculas resultantes. d) Transposiciones. A veces aparecen en el espectro de masas fragmentos cuya presencia no se puede explicar por la simple ruptura de enlaces y que son consecuencia de procesos de transposición. Muchos de estos procesos tienen lugar a través de mecanismos específicos que se conocen suficientemente bien como para que los iones resultantes sean útiles para la determinación de estructuras. La fuerza determinante de estos procesos es la mayor estabilidad de las especies que se forman. Uno de los más importantes es el debido a la migración de un átomo de hidrógeno de una parte a otra de una molécula. En la transposición de McLafferty interviene un sistema de electrones π y es común en olefinas, cetonas, ésteres, amidas, ácidos, etc. También son comunes las transposiciones en las que intervienen migraciones 1,2. En la interpretación de los espectros de masas existen los denominados picos metaestables que son muy útiles para elucidar mecanismos de descomposición. Un ion metastable es aquel que se descompone después de una aceleración casi completa en la fuente iónica pero antes del análisis completo de las masas. El ion producido da lugar a un pico algo difuso en el espectro por debajo de su masa llamado “pico metastable” (m*) y representa realmente el producto de la descomposición de un ion metastable. En general, aunque no siempre, los iones origen (m1) y producto de esta descomposición (m2) son también visibles a su m/e real en el espectro. El valor de m* se puede calcular mediante la siguiente fórmula: m* = m22/m1, lo que permite relacionarlo con los valores de m1 y m2. Sección experimental Reactivos y disolventes Seis compuestos orgánicos problema. Material Espectrómetro de masas, viales. Habitualmente los espectrómetros de masas son manejados por personal especializado y el químico orgánico se limita al proceso de interpretación de los espectros. Se pueden analizar muestras sólidas, líquidas o gaseosas, así como mezclas o sustancias puras. Dependiendo del tipo de muestra existen varios sistemas de introducción. La más común es la inserción directa aunque también es habitual la introducción a través de un cromatógrafo de gases, lo que permite analizar mezclas. Además, se debe lograr la entrada de la fuente iónica de un flujo relativamente constante de muestra vaporizada durante el barrido del espectro. Los gases y líquidos que poseen presión de vapor elevada a temperatura ambiente pueden introducirse en frío. En el caso de líquidos o sólidos no volátiles pero relativamente estables térmicamente, se utiliza el mismo sistema pero en caliente. Si la muestra es inestable térmicamente, o tiene presión de vapor muy baja a esas temperaturas, se introduce directamente en la cámara de ionización en el extremo de una sonda que puede calentarse. 1.4 Difracción de Rayos X Esta técnica nos permite obtener la estructura exacta de la molécula en estado sólido así como su disposición en la red cristalina. Sin embargo, para realizar un análisis por difracción de Rayos X es imprescindible obtener un monocristal del compuesto problema. El método más simple para obtener monocristales consiste en la evaporación lenta y a temperatura ambiente de una disolución saturada de la muestra. Esto se consigue dejando abierto el matraz de cristalización en vitrina, sobre todo si el disolvente es tóxico. Otro método se basa en la difusión lenta de dos disolventes: uno más denso (el disolvente en el que se disuelve la muestra, habitualmente diclorometano) y otro menos denso (el disolvente en el que la muestra es menos soluble, éter etílico o hexano). El procedimiento experimental comienza con la disolución de la muestra en un pequeño vial lo más estrecho posible. Después se vierte con cuidado un volumen aproximadamente semejante del disolvente menos denso, se cierra el vial y se espera a que los disolventes se mezclen. Es esencial que la adición del segundo disolvente se haga con lentitud para que la interfase permanezca perfectamente clara. La difusión de ambos disolventes provocará la formación de cristales en la interfase. Otra opción es colocar el vial con la disolución de la muestra en otro cristalizador más grande al que se adiciona un disolvente más volátil, en el que el compuesto es insoluble. El sistema se tapa y se deja en reposo. El disolvente más volátil irá condensando en el vial interior y tras la mezcla de las interfases se producirá la formación de cristales. Un tercer método, es la cocristalización con óxido de trifenilfosfina, sobre todo para compuestos orgánicos que dan cristales de mala calidad o sólo en pequeñas cantidades pero no se describirá aquí ya que excede los objetivos de este curso. 1.5 Aplicación a ejemplos sencillos Realice los espectros de IR, RMN de protón y carbono y EM de los compuestos obtenidos en el resto de prácticas, preparando las muestras según se ha descrito anteriormente y lleve a cabo la determinación de las estructuras correspondientes. Ha de tener en cuenta que, primero se registra el espectro de IR; luego el RMN de protón y, sin cambiar la muestra, obtenga el espectro de RMN de carbono, lo que permite ahorrar tiempo ya que el aparato estará optimizado. Asegúrese de imprimir, además del espectro, los desplazamientos químicos, los valores de las integrales y cualquier multiplete reconocible, expandiéndolo si es necesario para que se distingan los picos y se puedan medir las constantes de acoplamiento.