to get the file
Anuncio

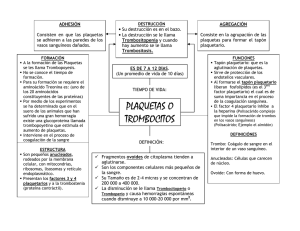
Facultad de Medicina Departamento de Fisiología FISIOLOGÍA HUMANA BLOQUE 4. HEMATOLOGÍA Tema 16. Hemostasia y coagulación sanguínea Prof. Miguel García Salom E‐mail: mgsalom@um.es. Tlfno. 868 88 3952 Facultad de Medicina. Despacho B1.1.037. Campus de Espinardo HEMOSTASIA “Proceso fisiológico que impide las pérdidas fi i ló i i id l é did de sangre por los vasos pequeños” ( (Se activa cuando se daña el endotelio vascular). i d d ñ l d li l ) Comprende cuatro mecanismos: 1. Vasoconstricción y presión tisular yp 2. Formación del tapón plaquetario 2. Formación del tapón plaquetario (H. Primaria) 3. Coagulación de la sangre (H.Secundaria 3. Coagulación de la sangre ( H.Secundaria) ) 4. Fibrinolísis 4. Fibrinolísis VASOCONSTRICCIÓN Y PRESIÓN TISULAR Mecanismos 1. Espasmo miogénico local iniciado por el daño directo de la pared arterial 2. Liberación de autacoides locales procedentes de los tejidos traumatizados y de las plaquetas: Tromboxano A2 plaquetario. Muy importante en vasos pequeños. 3. Respuesta nerviosa local, debida al dolor y a otros impulsos sensoriales A mayor trauma vascular, mayor grado de espasmo vascular y mayor d duración del mismo (desde minutos a horas) ió d l i d d i h HEMOSTASIA “Proceso fisiológico que impide las pérdidas de sangre por los vasos pequeños” (Se activa cuando se daña el endotelio vascular). (Se activa cuando se daña el endotelio vascular) Comprende cuatro mecanismos: 1. Vasoconstricción y presión tisular V i ió ió i l 2. Formación del tapón plaquetario 22. Formación del tapón Formación del tapón plaquetario Formación del tapón plaquetario (H. Primaria)) (H. Primaria 3. Coagulación de la sangre (H.Secundaria 3. Coagulación de la sangre ( H.Secundaria) ) 4 Fibrinolísis 4. Fibrinolísis 4. Fibrinolísis PLAQUETAS Proceden de los megacariocitos. Cada megacariocito da lugar a una media de 50 plaquetas/día. Tamaño: 2 3 µm de diámetro y una vida media: Tamaño: 2‐3 µm de diámetro y una vida media: 5‐9 días. Sin núcleo. Liberan numerosos factores de crecimiento, incluyendo al factor de crecimiento plaquetario (PDGF), un potente agente quimiotáctico, y el TGF‐β (estimula la deposición de matriz extracelular). Estos 2 factores de crecimiento tienen una importante factores de crecimiento tienen una importante participación en la reparación y regeneración tisular. Otros factores de crecimiento beneficiosos producidos por la plaqueta son el FGF, el IGF‐1. Otros factores: VEGF. PLAQUETAS Estructura Zona periférica: Membrana con 1.Sistema canalicular conectado a la superficie (SCCS), y 2. Sistema tubular denso, ‐estrechamente alineado con el anterior‐ que secuestra Ca2+ y tiene enzimas para la activación anterior‐, que secuestra Ca plaquetaria. Zona central: gránulos α : Se funden con el SCCS durante la activación plaquetaria. : Se funden con el SCCS durante la activación plaquetaria Contienen proteínas sintetizadas (PDGF, PF‐4, HMWK, Fibronectina, Trombospondina, etc) o absorbidas del plasma (albúmina, fibrinógeno, vWF, FV, etc). δ (cuerpos densos): 2‐7/plaqueta. Al activarse migran a la membrana plasmática y liberan su contenido directamente. Contienen ADP, ATP, Serotonina, Ca2+, Mg2+, COX, etc Receptores de membrana Grupo de glicoproteínas que sirven de receptores a factores de coagulación y moléculas de adhesión (GpIb:FvW), Gp IIb/IIIa: g y p VI) Fibrinógeno y Gp http://upload.wikimedia.org/wikipedia/commons/thumb/5/5f /Giant platelets JPG/250px Giant platelets JPG /Giant_platelets.JPG/250px‐Giant_platelets.JPG PLAQUETAS Funciones: = Forman tapones hemostáticos = Activan proteínas coagulación p g = Producen mediadores que reparan el vaso Agregación g egac ó de de plaquetas p aquetas e en un frotis u ot s sa sanguíneo. gu eo Número: 150‐300.000/μL Distribución: 2/3 circulantes 1/3 reserva esplénica Vid di dí Vida media: 5‐9 días (destrucción en bazo por macrófagos) http://upload.wikimedia.org/wikipedia/commons/thumb/8/89/Platelets.jpg/ 220px‐Platelets.jpg 2. FORMACIÓN DEL TAPÓN PLAQUETARIO Pl Plaqueta inactiva t i ti Pl Plaqueta activada t ti d http://www.reddymed.com/hdbc_platelet_overview.htm http://www reddymed com/hdbc platelet rinhemo htm http://www.reddymed.com/hdbc_platelet_rinhemo.htm FORMACIÓN DEL TAPÓN PLAQUETARIO Copyright © 2007 Pearson Education, Inc., publishing as Benjamin Cummings HEMOSTASIA “Proceso fisiológico que impide las pérdidas oceso f s o óg co que p de as pé d das de sangre por los vasos pequeños” ( (Se activa cuando se daña el endotelio vascular). ) Comprende cuatro mecanismos: 1. Vasoconstricción y presión tisular V i ió ió i l 2. Formación del tapón plaquetario (H. Primaria) 3. Coagulación de la sangre (H.Secundaria) (formación de una red de fibrina que refuerza el trombo plaquetario) 4. Fibrinolísis 4. Fibrinolísis 3 3. FORMACIÓN DEL COÁGULO Hay más de 50 sustancias en sangre y tejidos que activan o inhiben la y 5 g y j q coagulación de la sangre. a. Pared vascular intacta: Predominio anti‐coagulante b. Rotura pared vascular: Predominio pro coagulante b. Rotura pared vascular: Predominio pro‐coagulante Mecanismo general de formación del coágulo: Se produce en 3 etapas: 1ª Formación del activador de la protrombina 1ª. Formación del activador de la protrombina. 2ª. Conversión de protrombina en trombina, proceso que necesita calcio 3ª. Transformación del fibrinógeno en fibrina, atrapando a ª T f ió d l fib i ó fib i t d plaquetas, células sanguíneas y plasma. La produce la trombina polimerizando moléculas de fibrinógeno en fibras de fibrina. Es un proceso que se retroalimenta a si mismo (la trombina activa a la protrombina y a otros factores de la coagulación) 3 3. FORMACIÓN DEL COÁGULO Rotura vascular 15‐20 s/1‐2 min Activadores vasculares, plaquetarios y plasmáticos 3‐6 min 6 i C á l (fib i l Coágulo (fibrina, plaquetas, céls. sanguíneas y plasma) t él í l ) Factor estabilizador de la fibrina Estabilización del coágulo 20 min‐1 h Retracción del coágulo (requiere plaquetas suficientes que liberan factor estabilizador de la fibrina) Invasión por fibroblastos Disolución del coágulo Formación de tejido conjuntivo FACTORES DE LA COAGULACIÓN Factor Sinónimos Características F. I Fibrinógeno Pm = 340.000. Formado por 3 dímeros α, β y γ en hígado F. II Protrombina Pm = 70.000. Síntesis hepática dependiente de Vit. K F. III Factor tisular Tromboplastinas tisular (complejo de Lipoproteínas y colesterol) Calcio F. V Proacelerina F. VII Proconvertina F VIII F. VIII Factor antihemofílico A Dos partes: F VIII coagulante o F VIIIc (Hemofilia A) y F VIII de von Willebrand (plaquetas) F. IX Factor antihemofílico B Síntesis hepática dependiente de Vit. K F. X Factor Stuart F XI F. XI Factor antihemofílico C F. XII Factor de Hageman F. XIII Factor estabilizador de la fibrina Factor Fletcher Precalicreína Factor Fitzgerald Kininógeno de alto Pm Antitrombina III Proteína C í C Proteína S Pm = 45.000. Síntesis hepática dependiente de Vit. K Muy importante por actuar en las 2 vías. Síntesis hepática dependiente de Vit K Dímero de Pm = 160 000 Antecedente plasmático de la tromboplastina Dímero de Pm = 160.000. Antecedente plasmático de la tromboplastina 3 3. FORMACIÓN DEL COÁGULO Hall: Guyton y Hall.Tratado de Fisiología Médica 12ªEd. Copyright©2011 Elsevier Saunders 3 3. FORMACIÓN DEL COÁGULO Esquema de la conversión de protrombina en trombina y de la q p y polimerización del fibrinógeno en fibrina 3 3. FORMACIÓN DEL COÁGULO La coagulación por la vía L l ió l í extrínseca no se produce sin calcio Hall: Guyton y Hall.Tratado de Fisiología Médica 12ªEd. Copyright©2011 Elsevier Saunders 3 3. FORMACIÓN DEL COÁGULO Hall: Guyton y Hall.Tratado de Fisiología Médica 12ªEd. Copyright©2011 Elsevier Saunders 3 3. FORMACIÓN DEL COÁGULO La coagulación por la vía intrínseca no se produce d sin i calcio l i Factores anti-hemofílicos Hall: Guyton y Hall.Tratado de Fisiología Médica 12ªEd. Copyright©2011 Elsevier Saunders 3 3. FORMACIÓN DEL COÁGULO 1. Lesión endotelial 4. Vasoconstricción 2. Adhesión plaquetaria 5. Agregación plaquetaria 3. Activación plaquetaria 6. Formación del trombo Ver animación en http://www.reddymed.com/hdbc_overview.htm 3. FORMACIÓN DE LA RED DE FIBRINA http://en.wikipedia.org/wiki/Fibrin http://commons.wikimedia.org/wiki/File:Fibrina.jpg?uselang=es Filamentos azules de fibrina atrapan hematíes (rojo) y plaquetas (rosa) para formar un coágulo. http://sandwalk.blogspot.com.es/2008/04/fibrin‐and‐blood‐clots.html RESUMEN DE LA COAGULACIÓN FL: Fosfolípidos plaquetarios Factor XIII: Factor estabilizador de la fibrina Modificado de http://es.wikipedia.org/wiki/Coagulaci%C3%B3n HEMOSTASIA “Proceso Proceso fisiológico que impide las pérdidas de sangre por los vasos pequeños” (Se activa cuando se daña el endotelio vascular) vascular). Comprende cuatro mecanismos: 1. Vasoconstricción y presión tisular 2. Formación del tapón plaquetario (H. Primaria) 3. Coagulación de la sangre (H.Secundaria) 4. Fibrinólisis FIBRINOLISIS * * * La plasmina L l i también degrada a fibrinógeno, protrombina y a los factores V, VIII y XII bié d d fib i ó bi l f V VIII XII PDFs: Productos de degradación de la fibrina RESUMEN DE LA HEMOSTASIA Y COAGULACIÓN PDFs: ESTUDIO DE LA COAGULACIÓN 11.‐Recuento de plaquetas: atención a los falsos positivos (adhesión). ‐Recuento de plaquetas: atención a los falsos positivos (adhesión) Estudio de la agregación plaquetaria (ADP + colágeno). 2.‐ Fragilidad o resistencia capilar (Prueba de Rumpel‐Leede): g p p más de 6 petequias. 3.‐ Tiempo de hemorragia (prueba de Ivy): Comprobamos la formación del tapón de plaquetas (hemostasia), (incisión en antebrazo y presión de 40 mmHg). Máximo de 7 minutos. 4 ‐ Tiempo de protrombina (TP): estudio de la vía extrínseca. Añadir 4.‐ Tiempo de protrombina (TP): estudio de la vía extrínseca Añadir al plasma tromboplastina y calcio. Se mide en segundos (12”‐15”) o en porcentajes con respecto al suero control 5.‐ Tiempo de tromboplastina parcial activado (TTPA): estudio de la vía intrínseca. Se añade cefalina y kaolin. Normal: 30”‐ 40”. COAGULOPATÍAS Enfermedades que cursan con hemorragias: 1. Enfermedades hepáticas: Hepatitis, cirrosis y atrofia aguda amarilla. 2. Enfermedades por déficit de Vitamina K. Cursan con déficit de los factores vitamino‐K dependientes: factores II, VII, IX y X. También déficit de la p , , y Proteína C 3. Hemofilias: Déficit hereditario de los factores VIII (hemofilia A ‐85%) o IX (hemofilia B 15%). o IX (hemofilia B ‐15%) 4. Trombocitopenias. Se producen por insuficiente número de plaquetas. Enfermedades tromboembólicas: 1. Producidas por alteración del endotelio: Arteriosclerosis, infecciones o traumatismos i 2. Producidas por estasis sanguínea: Trombosis venosas 3. Producidas por infecciones: Septicemias, lipopolisacárido.