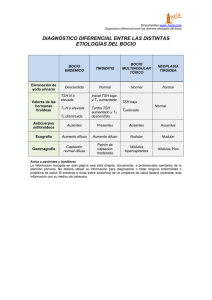
casos clínicos - RAEM | Revista Argentina de Endocrinología y
Anuncio

Revista Argentina de Endocrinología y Metabolismo Copyright 2012 por la Sociedad Argentina de Endocrinología y Metabolismo FASEN 2012 CASOS CLÍNICOS IX CONGRESO FASEN Viernes 5 de octubre Vol 49 Nº Supl. CASOS CLÍNICOS 123 - Tiroides 1 - 1 a 8 - Salón Castellanos I - Coordinador: AM Orlandi APARICIÓN DE GINECOMASTIA UNILATERAL EN UN GERONTE LUEGO DE LA ADMINISTRACIÓN DE DOMPERIDONA Mingote E, Rosmarin M, Fossati MP, Faingold MC, Pacenza NA. 1 Servicio de Endocrinología y Metabolismo. Unidad Asistencial “Por más Salud” “Dr. César Milstein” CABA, Argentina. La ginecomastia, agrandamiento mamario en el hombre, es de origen multifactorial. La etiología medicamentosa es una de las causas más frecuente. La domperidona, antagonista dopaminérgico que no cruza libremente la barrera hémato-encefálica, muy utilizada en el arsenal terapéutico de la patología gastroenterológica, puede ocasionalmente producir una leve hiperprolactinemia, pero muy raramente fue reportada como causal de ginecomastia. Se presenta el caso de un paciente de 76 años derivado por agrandamiento y dolor de mama izquierda, de 2 meses de evolución. En cuanto a sus antecedentes personales presentaba diabetes tipo 2, hipertensión arterial, gastritis crónica y arteriopatía periférica. Cirugía de cáncer de próstata hacía 15 años, no habiendo recibido hormono ni radioterapia. Antecedente de parotiditis sin orquitis y negaba antecedentes de infección o traumatismo testicular y/o mamario. Medicación al momento de la consulta: gliclazida 60 mg/d, losartán 100 mg/d, indapamida 1,5 mg/d, omeprazol 20 mg/d, domperidona 20 mg/d, lorazepam 2 mg/d, cilostazol 200 mg/d. El paciente no presentaba antecedentes familiares de ginecomastia, tiroideopatía ni de diabetes. Traía una ecografía mamaria que revelaba una formación sólida de 13 x 9 mm a nivel retroareolar izquierdo, sin alteraciones en mama derecha. Al examen físico: Talla: 1,69 m, Peso: 64,7 kg. Tiroides palpable normal. Testículos pequeños y sensibles (8 ml Prader). Varicocele bilateral. Mamas: palpación de tejido retroareolar izquierdo sensible a la palpación, de 1,5 a 1 cm de diámetro, sin galactorrea a la expresión. El laboratorio inicial mostró una Testosterona total (TT) y biodisponible (Tbio) bajas, LH y FSH elevadas, un Estradiol (E2) normal y una prolactina (PRL) de 71,2 ng/mL. Los marcadores tumorales (AFP y β-HCG) fueron negativos y la LDH estaba dentro del rango de referencia. Se le indica solamente la suspensión de la domperidona. A los 40 días, ya no presentaba dolor mamario espontáneo ni al tacto y se percibía un achicamiento del botón retroareolar. A los 90 días se constata desaparición del botón mamario retroareolar izquierdo, coincidiendo con el descenso de los valores de PRL. En el control al año de la suspensión de la domperidona el paciente estaba asintomático, sin botón mamario palpable y con ecografía mamaria normal. La evolución de los valores hormonales se refleja en la siguiente tabla: Basal (Mayo 2011) A los 12 días de susp. de la DOMP A los 60 días de susp. de la DOMP Al año de la susp. de la DOMP Valores de referencia Método TT 2,40 3,40 3,26 2,02 2,9 a 8,2 ng/mL QL Tbio 0,69 0,56 0,70 1,5 a 6,5 ng/mL Fórmula Vermeulen LH 16,7 21,5 1,7 a 8,6 mUI/mL QL FSH 64,8 67,0 1,5 a 13 mUI/mL QL PRL 71,2 29,0 3 a 18 ng/mL QL E2 <20 <20 Hasta 55 pg/mL QL 17,3 36,7 16,2 Conclusiones: Se presenta el caso de un hombre de 76 años portador de una ginecomastia izquierda con hiperprolactinemia secundaria a la ingesta de domperidona, que remitió dicho cuadro con la suspensión del fármaco. Si bien es conocida la asociación de domperidona e hiperprolactinemia, hemos hallado solo 2 casos reportados en la bibliografía (un adulto y un niño) de presentación de ginecomastia en relación con la ingesta de domperidona(1,2). 1. Keating JP et al., Postgrad Med J 67 (786):401-2, 1991. 2. van Der Steen M et al., Lancet 2(8303):884-5, 1982. RAEM 2012. Vol 49 Nº Supl. 124 2 METÁSTASIS INTRATIROIDEAS: PRESENTACIÓN DE TRES CASOS Moncet D*, Zoppi J**, Isaac G*. *Servicio de Endocrinología y **Patología. Hospital Privado de Comunidad.Mar del Plata danmoncet@gmail.com Córdoba 4545. Tel. 223-4990000. Mar del Plata, Argentina. Las metástasis intraparenquimatosas de la tiroides (MIT) representan el 1,4-3% de las neoplasias y entre el 0,05 y 0,1% de todas las enfermedades de tiroides. Su incidencia en autopsias es mayor llegando al 10% o más. Su observación en la práctica es excepcional. Los tumores que más frecuentemente dan metástasis tiroideas son el melanoma cutáneo, carcinoma de células claras de riñón, el cáncer de pulmón y mama; y menos frecuente los originados en aparato digestivo. Las MIT pueden semejar tumores primarios de la glándula, pudiendo ser muy difícil el diagnóstico preoperatorio tanto del punto de vista clínico, como en los estudios de diagnóstico por imágenes, en la punción con aguja fina (PAAF) e incluso en el estudio de la pieza operatoria. Nuestro objetivo es describir 3 pacientes con MIT de tumores primarios localizados en riñón, mama y cólon, que se presentaron como un bocio y fueron remitidos al Servicio de Endocrinología. Se revisó el registro de patología nodular tiroidea del Servicio y se seleccionaron los pacientes que presentaron MIT. Paciente 1: Mujer de 79 años con antecedentes 3 años previos a la consulta de un carcinoma de células claras del riñón izquierdo con invasión de la vena renal, vía excretora y metástasis en ganglios precavos derechos y otro carcinoma de igual histologia en el riñón derecho, resecados quirúrgicamente. Al año, presentó en una TAC un nódulo pulmonar de 1,5 cm y un nódulo de 4 cm en LD tiroideo que desplazaba la carótida. En la ecografía tiroidea se observó un nódulo de 4,8 x 3,6 cm heterogéneo de contornos parcialmente definidos con numerosos vasos internos. La TSH fue de 6,8 mU/l y la T4 de 7,9 ug/dl Se realizó una hemitiroidectomía derecha y en el estudio intraoperatorio se diagnosticó una metástasis de carcinoma de células claras del riñón que fue confirmado en el estudio diferido. La paciente evolucionó tórpidamente con compromiso multisistémico, falleciendo a los 4 años del diagnóstico del nódulo tiroideo. Paciente 2: Mujer de 63 años con antecedentes 2 años previos a la consulta de carcinoma ductal infiltrante con metástasis ganglionares axilares de mama izquierda con tratamiento quirúrgico, quimio y radioterapia y tamoxifeno. En el momento de la consulta con el Servicio presentó una hemiparesia braquiocrural derecha por metástasis cerebral; y disfonía con disnea por parálisis bilateral de las cuerdas vocales secundaria a la invasión de un tumor tiroideo palpable al exámen. La TSH fue de 4,3 mU/l. Se le realizó traqueostomía y biopsia del tumor tiroideo que se diagnosticó como metástasis de carcinoma ductal de la mama. Murió con enfermedad diseminada 30 días después. Paciente 3: Mujer de 86 años con antecedentes de carcinoma avanzado de cólon en mal estado general que presentó un bocio difuso a la palpación con una ecografía que mostró un bocio multinodular con adenopatías bilaterales e infiltración del músculo esternocleidomastoideo derecho. Se le realizó PAAF del nódulo dominante en LD que se diagnosticó como metástasis de adenocarcinoma compatible con un origen en cólon/recto. La paciente murió 2 meses después de la evaluación. Conclusión: Presentamos 3 casos poco frecuentes de bocio, con un promedio de edad ligeramente mayor al reportado y con predominio de mujeres. La presencia de MIT fue observada en estadios avanzados de la enfermedad de base. En 2 de ellos se pudo documentar la aparición entre los 2-3 años del inicio de la neoplasia, aunque pueden aparecer hasta 10 años posteriores. El diagnóstico puede ser dificultoso principalmente si no se conoce el antecedente del tumor primario. Debe tenerse en cuenta la posibilidad de MIT en pacientes añosos, con antecedentes neoplásicos y con citologías o histologías no habituales o con tumores poco diferenciados. FASEN 2012 CASOS CLÍNICOS TAPONAMIENTO CARDÍACO COMO DEBUT DE CARCINOMA PAPILAR DE TIROIDES VARIEDAD ESCLEROSANTE DIFUSA Bertolino ML, Wyse EP, Wior ME, Yaniskowski ML, Giovannini A, Ramacciotti CF, Mendez AM. Servicio de Endocrinología, Hospital Privado, Córdoba, Argentina. 125 3 Introducción: La variedad esclerosante difusa del carcinoma papilar de tiroides (CPT) es una entidad infrecuente (2-6 % de los CPT). Se observa predominantemente en mujeres jóvenes, la mayoría con metástasis ganglionares al momento del diagnóstico, y también son comunes las metástasis pulmonares. Objetivo: presentar el caso de una paciente con carcinoma papilar variante esclerosante difusa que debutó de manera inusual y evolucionó en poco tiempo de manera fatal. Materiales y Métodos: se trata de una mujer de 36 años, sana previa, con antecedentes de su madre con cáncer de mama y su padre de pulmón. Ingresa al hospital por taponamiento cardíaco. Se le realizó una pericardiocentesis con ventana pericárdica, donde se observó un nódulo pulmonar, el cual fue resecado. La anatomía patológica informó metástasis (mtts) de carcinoma papilar con margen focalmente comprometido, pericardio y pleura parietal con inflamación leve y fibrosis. CK7 (+) TIF 1 (+) Vimentina (+) Tg (+) focal. Se sugirió descartar primario de tiroides. Se solicitó el siguiente laboratorio: T4: 8.7 ug/dl, TSH: 4.19 uU/ml, ATPO: 10 UI/ml, ATG: <10 UI/ml, Tg: 10.3 ng/ml. La ecografía tiroidea informó: glándula de dimensiones normales y contornos regulares; ecoestructura homogénea sin lesiones focalizadas, con múltiples imágenes entre 8 y 13 mm, compatibles con adenopatías inespecíficas en región anterior y medial de vasos carotídeos. Doppler negativo. Un estudio PET mostró captación difusa en parénquima pulmonar y adenopatías laterocervicales. Se derivó a nuestro servicio donde se solicitó PAAF de ganglios cervicales. La misma fue positiva para carcinoma papilar. Se decidió realizar una tiroidectomía total con vaciamiento ganglionar cervical bilateral, durante la cual no se hallaron nódulos macroscópicos. Resultados: la AP informó Carcinoma Papilar Esclerosante Difuso bilateral de tiroides, con áreas sólidas, 1 ganglio supraclavicular izquierdo con metástasis y 16 ganglios en cadena yugulocarotídea derecha e izquierda con mtts. Se realizó dosimetría y se indicaron 300 mCi de Yodo 131. El laboratorio previo fue: TSH: 73.18 uU/ml, Tg: 7.9 ng/ml, ATG: 17 UI/ml. El BCT mostró captación en región cervical, en ambos parénquimas pulmonares a predominio izquierdo y 1/3 superior de fémur derecho (T3N1M1). La paciente reingresó al hospital en varias oportunidades con derrame pleural bilateral y pericárdico. Presentó trombosis en 2 venas yugulares internas, TVP en miembros inferiores y TEPA derecho. Se realizaron pleurocentesis evacuadoras, con mala evolución clínica. El Servicio de Oncología sugirió tratamiento quimioterápico con Carboplatino + Paclitaxel. Se realizaron 2 ciclos. Siguió con evolución poco favorable. En la última internación se le realizó una toracotomía exploradora con ventana pericardio-peritoneal. La AP del líquido pericárdico y pleural informó citología maligna vinculable a carcinoma papilar y pericardio infiltrado por carcinoma papilar vinculable a primario de tiroides. Paciente muy inestable, fallece a los 6 meses del debut de su enfermedad con taponamiento cardíaco. Discusión: la variante esclerosante difusa del CPT se presenta característicamente con compromiso rápido y difuso de la glándula y clínicamente simula una tiroiditis linfocitaria crónica o un linfoma. Ecográficamente puede presentar dificultades diagnósticas. Algunos autores refieren que tiene peor pronóstico que la variante clásica del CPT; sin embargo revisiones más recientes sugieren que el pronóstico sería comparable con el CPT clásico de alto riesgo. La razón por la cual presentamos este caso fue motivada por la baja frecuencia con la que se observa este tipo de tumor y la evolución rápidamente fatal que presentó nuestra paciente. RAEM 2012. Vol 49 Nº Supl. 126 4 TIROIDITIS INFECCIOSA POR BACILO DE KOCH Luna M, Barrera Olarte N, Figueroa AM, Noguera MA Hospital Ángel C. Padilla. Alberdi 550, Tucumán, Argentina. Introducción: La tuberculosis primitiva de la glándula tiroidea es rara. En el IPT desde 1951 se han observado dos casos. Se trata, generalmente, de la expresión local de una diseminación miliar, o bien de una forma fibrocaseosa. El diagnóstico sólo se afirma si se logra aislar el germen causal o por la histología. Se debe efectuar tratamiento específico y, si se trata de una forma fibrocaseosa, cirugía. Caso clínico: Paciente de 21 años, que consultó por traumatismo máxilofacial secundario a accidente de tránsito, donde indican tratamiento con Corticoides que la paciente utilizo en tiempo prolongado. Luego de dos meses presentó tumoraciones múltiples en región facial y cervical; identificándose adenopatías cervicales de forma redondeada, situadas en región submaxilar y cervicolateral. Se realizó PAAF de Tiroides, Ganglio Linfático y Áreas Fluctuantes que informaba: extendido hemorrágico con abundantes leucocitos PMN, cuadro histopatológico compatible con Tiroiditis Aguda Abscedada y Linfadenitis supurativa. El cultivo para gérmenes comunes y micológico arrojó resultados negativos. La paciente evoluciona con mal estado general, se interna en Sala de Clínica Médica. Se realiza nueva PAAF Tiroidea y de Adenopatías Cervicales y en la tinción del ganglio informa BAAR (+) el laboratorio demostró función tiroidea normal, con ATPO y Atg negativos. Se instaura tratamiento para TBC ganglionar con afectación tiroidea con Triple asociación más Etambutol. Se realizó toilette cervical extensa, biopsias ganglionares y de tejido. El cultivo de la secreción obtenida dió resultado BAAR positivo (++) en ganglio. Discusión: La Tuberculosis es la enfermedad infectocontagiosa más difundida en el mundo. La TBC tiroidea es una localización sumamente infrecuente. En 1893, describió el primer caso de Tuberculosis Tiroidea (TT) sin evidencia de compromiso pulmonar; en el estudio microscópico de 20.758 tiroidectomías realizadas en la Clínica Mayo, en un período de 11 años, se diagnosticaron 21 Tuberculosis (0,1 % del total). Se observó que algunos casos de TBC tiroidea han simulado adenocarcinoma de la glándula Tiroides en el informe anatomopatológico de la PAAF, haciéndose el diagnóstico definitivo y correcto de TBC tiroidea por medio de la biopsia glandular pos tiroidectomía. El objetivo de este reporte es la presentación de una forma infrecuente de tuberculosis extrapulmonar, resaltando la importancia de realizar diagnóstico diferencial con el carcinoma papilar de tiroides por medio de métodos conservadores como la PAAF y recurrir al tratamiento farmacológico de la TBC para evitar la tiroidectomía, obteniendo buenos resultados terapéuticos con drogas antituberculosas en una paciente con tiroiditis infecciosa crónica eutiroidea. Conclusión: Paciente que secundariamente al uso de Glucocorticoides desarrolló la activación de TBC primaria, presentando TBC Ganglionar y Tiroiditis Aguda Abscedada por Bacilo de Koch sin alteración de la función tiroidea pero con alteración estructural e infecciosa de la glándula; se realizó tratamiento con drogas antituberculosas y cirugía que permitió la toma de muestra del material para confirmación del diagnóstico y también mitigar la sintomatología de la paciente. Vemos que a pesar de los múltiples esfuerzos epidemiológicos para evitar esta antigua enfermedad puede presentarse en casos tan inverosímiles como ser una TBC Tiroidea, que gracias a la “nobleza” de esta glándula la función de la misma se presentó inalterada y al entrenamiento tanto médico como quirúrgico pudo preservársela, a diferencia de otros casos descriptos. Bibliografía 1. 2. 3. Kang M, Ojili V, Khandelwal N. Primary tuberculosis of the thyroid gland. Hormones (Athens). 2008; 7:330-3. Bulbuloglu E, Ciralik H, Okur E. Tuberculosis of the thyroid gland: review of the literature. World J Surg 2006; 30:149-55. Raimondo y Col. Enfermedades Infecciosas. UNT 2010; 22:212-13 FASEN 2012 CASOS CLÍNICOS METÁSTASIS DE CARCINOMA DIFERENCIADO DE TIROIDES EN SENO CAVERNOSO Concilio V1, Lowenstein A1, Latorre F2, Colobraro A3, Monteros Alvi M4, De Los Rios A4, Rogozinski A1 1 División endocrinología, 2Neurocirugía y 3Anatomía patológica Hospital JM Ramos Mejía. CABA, Argentina. 4Hospital Dr A. Oñativia. Salta, Argentina. veronicaconcilio@hotmail.com, Urquiza 609. CABA (1221). 127 5 Introducción: El carcinoma diferenciado de tiroides (CDT) suele tener buen pronóstico a largo plazo. Sin embargo las metástasis a distancia son un evento grave en estos pacientes. Los principales sitios de metástasis a distancia son pulmón y hueso. Existen otros sitos que debido a su baja incidencia se ignoran en la práctica clínica. Se presenta aquí un caso extremadamente raro de metástasis en seno cavernoso de CDT que se diagnostica 3 años posteriores al diagnostico inicial. Caso Clínico: Paciente de 61 años, oriundo de Salta. Derivado para estudio por presentar ptosis palpebral bilateral a predominio derecho y cefalea. Diagnóstico de CDT (12/2008). Con este hallazgo se realizó tiroidectomía total con vaciamiento ganglionar radical modificado derecho niveles II a VI. La anatomía patológica informó: LIzq nódulo de 2 cm: carcinoma papilar (CPT) no clásico patrón cribiforme, infiltración capsular y extratiroidea. LDer nódulo de 5x4 cm: carcinoma folicular (CFT) invasor con componentes de células claras. Metástasis de CPT de células altas en 2/4 ganglios cervicales. Recibió dosis de 150 mCi de I131. Posterior rastreo corporal total (RCT): positivo en cuello. Realizo nuevo control (9/2010) presentando ecografía tiroidea y RCT negativos, TSH 41 mUI/ml, TG<1 ng/ml, ATG 1756 UI/ml. Discontinuó seguimiento. En 12/2011 consulta nuevamente para control presentando compromiso neurológico por lo que es derivado para continuar estudio y tratamiento. Aporta: - RNM cerebro con gadolinio que informa formación sólida, en topografía selar y paraselar derecha de 24 x 26 x 17 mm que realza en forma homogénea con contraste y engloba en forma completa la arteria carótida intracavernosa extendiéndose en dirección anterior sobre el agujero óptico derecho. - Ecografía de cuello: negativa. TAC de tórax con imágenes nodulillares subpleurales izquierdas (sin cambios con respecto a TAC del 2008) - Laboratorio: TSH 0.56 mUI/ml, TG 5 ng/ml, ATG1366 UI/ml, bajo tratamiento con LT4 225ug/d (se aumenta dosis) Nuevos controles en el servicio: TSH<0.02 mUI/ml, TG 9.3 ng/ml, ATG 1473 UI/ml, PRL 13 ng/ml, cortisol 23 ug/dl, LH 5.6 mUI/ml, FSH 7.3 mUI/ml, Testosterona total 1.1 ng/ml, IGF1 158 ng/ml. Se realiza biopsia de lesión en seno cavernoso vía nasal endoscópica con anatomía patológica que informa: metástasis de cáncer papilar variante cribiforme. En estudio PET se observa recidiva local con secundarismo ganglionar, pulmonar y encefálico extraaxial. Foco hipermetabólico en seno cavernoso de 28 mm que produce lisis de estructuras óseas adyacentes. Se indica tratamiento con dosis terapéutica de 300 mCi I131, administrando TSH recombinante con corticoterapia pre y post dosis. Presentó RCP post dosis: “área hipercaptante del radiotrazador de tipo patológico en cabeza: zona centro derecha de base de cráneo, en cuello dos pequeñas áreas de captación en posición tiroidea, en mediastino superior varias formaciones hipercaptantes”. La terapia fue bien tolerada, sin presentar complicaciones. Comentario: La prevalencia de CPT con metástasis a distancia no supera el 1-2 %. Por otra parte la variante cribiforme es un subtipo histológico de muy baja frecuencia. En el caso de nuestro paciente, la imagen tumoral observada en la RNM asociado al antecedente de CPT y CFT orientaron hacia el diagnostico de metástasis de CDT en seno cavernoso. La biopsia confirmo el diagnostico y el estudio PET aportó la estadificación de la enfermedad y los datos anatómicos y metabólicos de la lesión. La localización de la metástasis es extremadamente inusual, con escasos reportes en la literatura. Consideramos que ante el hallazgo de un tumor en un paciente con antecedentes de CDT, las metástasis a distancia se deben plantear como diagnostico diferencial, aun tratándose de lugares poco usuales. RAEM 2012. Vol 49 Nº Supl. 128 6 BOCIO AMILOIDE Y LIPOMATOSO RECIDIVADO Cerioni V, Salvo Vázquez MC, Gonza N, Galíndez M, Monteros Alvi M, Cabrera J. Programa de Endocrinología, Sector Anatomía Patológica y Programa de Cirugía Del Hospital de Endocrinología y Metabolismo Dr. Arturo Oñativia. Paz Chaín 30 (4400) Salta, Argentina. mc_salvo@yahoo.com.ar Introducción: La amiloidosis es un grupo diverso de enfermedades caracterizada por depósito en el tejido extracelular de una proteína llamada amiloide. El bocio amiloide, es una rara entidad (0,045 %) definida por Von Eisselberg como el crecimiento de la glándula tiroides secundario al depósito abundante de esta proteína, que puede dar síntomas compresivos de las estructuras del cuello. Clínicamente puede simular una neoplasia por su crecimiento rápido y manifestarse con agrandamiento tiroideo gradual, difuso y firme. La infiltración subclínica es más común que la enfermedad sintomática. El diagnostico debe confirmarse por anatomía patológica. Caso Clínico: Paciente de sexo masculino de 46 años de edad que consulta por Bocio recidivado asociado a disfagia a sólidos en los últimos 15 días que mejora con la administración a glucocorticoides, sin desaparecer. Tiene antecedentes de: EPOC desde los 31 años, bronquiectasia e hipogamaglobunimemia congénita, vitiligo y antecedente de 2 cirugías de tiroides previas con neuroleptoanalgesia por neumopatía crónica que se realizaron en los años 1998: nodulectomía. A.P: adenoma folicular de tiroides y en el 2010 Tiroidectomía Subtotal Derecha. A.P: “Bocio amiloide y lipomatoso”. La biopsia de piel y tejido celular subcutáneo fue negativa para amiloide (Rojo Congo -) y con estudios de imágenes y laboratorio que no demostraron sospecha de compromiso sistémico al momento del diagnóstico. No regresa a control hasta el año 2012 donde consulta por síntomas compresivos ya descriptos. BMI 16,5, normotenso con TCS disminuido en forma generalizada. En cuello se ve cicatriz de tiroidectomía previa y glándula tiroidea que deforma el cuello, se palpan LD y LI aumentados de tamaño, consistencia firme, indoloros, móviles a la deglución y manualmente, superficie irregular, delimitándose borde inferior. Eutiroideo con ATPO 1/100 (+). Ecografía tiroidea: glándula con aumento difuso de tamaño, algunas imágenes quísticas con sedimento en su interior. Adenopatías (-) LD 30 x 66 x 29 mm; LI 40 x 67 x 30 mm. Captación y Centellograma: bocio difuso con captación de 6 %, 44 % y 49 %. TAC de cuello: (con cortes de 5 mm de ángulo mandibular a opérculo torácico) agrandamiento difuso de la glándula tiroides especialmente del LI, sin adenomegalias y laringe normal. Por síntomas compresivos, se decide realizar Tiroidectomía Total Bilateral con resultado histopatológico de Bocio Amiloide y Lipomatoso recidivado. Excelente evolución post quirúrgica con franca mejoría de los síntomas. Se inicia tratamiento sustitutivo con levotiroxina. Discusión: El compromiso de la glándula tiroides por amiloidosis primaria o secundaria corresponde en general a una infiltración difusa leve, que no determina aumento de tamaño glandular ni alteración funcional. La infiltración o depósito masivo de sustancia amiloide en tiroides ocasionado bocio es inusual y se asocia con lipomatosis como en nuestro caso. El estudio histológico permite confirmar la presencia de depósito de esta proteína de forma masiva y descartar neoplasias asociadas con el mismo (carcinoma medular) en pacientes con bocio sintomático compresivo. Ocasionalmente el bocio amiloide asintomático suele preceder a otras evidencias clínicas de amiloidosis sistémica como en el caso presentado donde se descartó compromiso sistémico hasta la fecha. Los síntomas locales del paciente se atribuyeron a su patología de base respiratoria en un principio pero luego se confirmó que los mismos se agravaban por la recidiva del bocio, mejorando luego de la Tiroidectomía Total. También cabe resaltar que dentro de su baja prevalencia se manifiesta generalmente como bocio difuso. FASEN 2012 CASOS CLÍNICOS COEXISTENCIA DE ENFERMEDAD DE GRAVES Y ESTRUMA OVÁRICO (DIAGNÓSTICO DIFERENCIAL DE HIPERTIROIDISMO REFRACTARIO) Pastorino Casas V, Borghi Torzillo MF, Capurro S, Lutfi RJ, Faure E. 129 7 Servicio de Endocrinología Hospital Churruca-Visca, CABA, Argentina. Introducción: En aquellos casos de hipertiroidismo refractarios luego de la Tiroidectomía Total o de la Radioablación se debería descartar focos de producción ectópica de hormonas tiroideas, ya sea por bocio endotorácico, metástasis de carcinoma tiroideo funcionante ó el estruma ovárico (ES). El ES es una patología poco frecuente que se observa en el 5 al 15 % de los tumores dermoides. Es una variante en la cual el tejido tiroideo constituye > 50 % del tumor. Representa 1 % de todos los tumores ováricos y el 2.7 % de todos los tumores dermoides. Tiene apariencia histológica que imita el adenoma tiroideo y las formas malignas se presentan más frecuentemente como tumores de mayor tamaño. La incidencia de hiperfunción tiroidea se observa en el 5-8 % de los pacientes, siendo infrecuente su asociación con Enfermedad de Graves. Nuestro objetivo es presentar un caso de esta rara asociación, diagnosticada por la persistencia de hipertiroidismo luego de la tiroidectomía total. Caso clínico: Mujer de 69 años de edad, con antecedentes de hipertiroidismo autoinmune tratado con drogas antitiroideas que frente a la persistencia se indicó Tiroidectomía total con Anatomía patológica de hiperplasia coloide y tiroiditis linfocítica. Consulta al mes de la cirugía para seguimiento de su patología tiroidea. Al examen físico, se presentaba clínicamente eutiroidea, con tiroides no palpable. Laboratorio sin tratamiento: TSH 0.15 uUI/ml (0,5-5), T4 libre 1.27 ng/dl (0.8-1.8). Centellograma tiroideo: Sin captación en cuello. Ecografía cervical: tiroides ausente sin adenomegalias. Laboratorio 60 días post quirúrgico: TSH 0.05 uUI/ml, T4 libre: 1.42 nd/dl, tiroglobulina 117 ng/ml, ATG 391.4 U/ml. Dado este resultado, se realiza TAC de cuello y tórax no visualizándose glándula tiroidea ni masa mediastinal. Ecografía abdominal y transvaginal: Imagen anecoica de aspecto quístico simple de 24 x 28 mm en ovario derecho. Ante la sospecha de tejido tiroideo ectópico y para descartar metástasis de carcinoma funcionante se solicita Rastreo Corporal Total con 10 mci de I131: Captación en resto cervical y región pelviana (línea media y hacia la derecha) de morfología esférica, supravesical, que coincide con topografía del quiste ovárico. TAC abdomen y pelvis que muestra masa en región anexial derecha de 92 x 72 x 12 mm de diámetro, contornos irregulares, heterogénea con múltiples calcificaciones con áreas de distinta densidad, que contacta con la pared anterior y lateral derecha del útero, desplaza al uréter pero no lo obstruye. Laboratorio: TSH < 0.01 uUI/ml, TRABS 18 % (VR: 12 %). Reinicia antitiroideos. Con diagnóstico presuntivo de estruma ovárico se realiza anexohisterectomía total. Anatomía patológica: Formación tumoral quística de ovario derecho constituida por tejido glandular tiroideo compatible con estruma ovárico, confirmando el diagnóstico. La paciente evoluciona favorablemente, laboratorio postquirúrgico: TSH 110 uUI/ml. Se inicia tratamiento sustitutivo con levotiroxina. Discusión: El estruma ovárico es causa poco frecuente de hipertiroidismo, en un alto porcentaje puede estar asociado a bocio multinodular y su principal diagnóstico diferencial son las metástasis de un carcinoma tiroideo. La asociación con Enfermedad de Graves es infrecuente. En aquellos pacientes con persistencia del hipertiroidismo luego de la tiroidectomía total o de la radioablación debería realizarse un scan corporal total con radioyodo para confirmar o excluir la presencia de estruma ovárico hiperfuncionante. 130 8 RAEM 2012. Vol 49 Nº Supl. LOCURA MIXEDEMATOSA: DIFICULTAD PARA EL SEGUIMIENTO DEL CARCINOMA PAPILAR DE TIROIDES Rosmarin MC, Morosán Allo YJ, Parisi C, Fossati MP, Rujelman R, Schnitman M, Brenta G, Faingold MC, Musso C. Servicio de Endocrinología y Metabolismo Unidad Asistencial “Por Más Salud” “Dr. César Milstein”, CABA, Argentina El hipotiroidismo puede estar asociado a múltiples desórdenes psiquiátricos entre los cuales psicosis, depresión y trastornos afectivos son los más frecuentes. La fisiopatología de la psicosis en el hipotiroidismo no está aún bien dilucidada. Aunque los episodios de manía están más relacionados al hipertiroidismo, el hipotiroidismo también puede manifestarlos. Si bien son pocos los casos descriptos en la literatura respecto a esta asociación, presentamos el caso de una paciente que luego de una tiroidectomía total por carcinoma diferenciado de tiroides, desencadenó un cuadro de psicosis atribuible a hipotiroidismo agudo severo. Caso clínico: Paciente de 62 años de edad que ingresó a la Guardia de nuestro Hospital con un cuadro de excitación psicomotriz de 24 horas de evolución. Como antecedentes personales presentaba HTA y una tiroidectomía total por carcinoma papilar de tiroides hacía 6 semanas. La paciente se encontraba sin tratamiento sustitutivo a la esperaba del dosaje de tiroglobulina estimulada y la dosis ablativa de iodo radioactivo. A 6 semanas de la tiroidectomía, comenzó a experimentar un comportamiento inapropiado con alucinaciones visuales y auditivas. El día previo a la consulta manifestó conductas auto y heteroagresivas hacia los miembros de su familia. Al examen físico presentaba una TA140/70mmHg, una FC 62 lpm y una Tº 36°C. Clínicamente se encontraba con facies abotagadas, macroglosia y edema bipalpebral. Ningún hallazgo patológico fue encontrado tanto en los exámenes de rutina de laboratorio, como en los estudios de imágenes (TC y RMN de encéfalo) y en el EEG. Fue evaluada por Servicio de Psiquiatría diagnosticándose cuadro de psicosis aguda. Se administró Haloperidol 2.5mg IM por única vez y VO además de Lorazepam 2.5mg/d y Levomepromazina 12.5mg/d VO. El perfil tiroideo de ingreso evidenciaba TSH 62.9mU/L, T4L <0.35ng/dL, T4 <1ug/dl y T3 70ng/dl. Se indicó Levotiroxina 200mcg en bolo EV y posteriormente tratamiento sustitutivo VO con una combinación de T3 20mcg/d y Levotiroxina 100mcg/d. Al cuarto día de tratamiento la paciente se encontraba globalmente orientada, sin conductas agresivas ni alucinaciones. Al finalizar la primer semana de tratamiento se realizó un nuevo perfil tiroideo que mostró TSH 39.4mU/L T4 10.4ug/dl, T4L 1ng/dl y T3 190ng/dl. Presentaba cierto grado de mejoría respecto al edema bipalpebral y facial. Fue dada de alta con Levotiroxina 150mcg/d. Luego de 15 días regresó para control por Consultorios Externos con TSH 16.1mU/L, T4 13.9ug/dl, T4L 1.8ng/dl y T3 90ng/dl. Se encontraba bajo tratamiento con drogas antipsicóticas cuyas dosis fueron reducidas progresivamente hasta la suspensión de las mismas. A los 6 meses de seguimiento, la paciente presentaba niveles hormonales en valores normales bajo TSH inhibida TSH <0.03mU/L, T4L 1.76ng/dl. En conclusión la psicosis puede ser la manifestación de un hipotiroidismo severo y debería tomarse en cuenta como diagnostico diferencial. En nuestro caso la paciente no tenía una base psiquiátrica predisponente y el único factor detonante fue el hipotiroidismo agudo. Si bien respondió favorablemente luego de una semana de tratamiento con drogas antipsicóticas, la rapidez de la recuperación y la falta de recurrencias en el seguimiento posterior podrían también atribuirse a la pronta reversión del hipotiroidismo con levotiroxina endovenosa. La dificultad planteada en el seguimiento del carcinoma papilar se encuentra en la posibilidad de nuevos episodios psiquiátricos durante la suspensión del tratamiento con levotiroxina. Como alternativa para evitar estos casos de hipotiroidismo severo con manifestaciones psiquiátricas importantes se podría recurrir al uso de TSH recombinante. FASEN 2012 Viernes 5 de octubre CASOS CLÍNICOS - Adrenal y Paratiroides - 9 y 16 - Salón Leguizamón - Coordinador: l. gomez 131 Lasalle HIPOGLUCEMIA INDUCIDA POR TUMOR ADRENAL Borghi MF, Saban M, Soutelo MJ, Leal M, Lutfi RJ. Servicio de Endocrinología Hospital Churruca- Visca, CABA, Argentina. 9 Introducción: Hipoglucemia se define como una glucemia plasmática inferior a 60 mg/dl y clínicamente presenta la triada de Whipple: signos y síntomas de hipoglucemia que revierten con glucosa oral o alimentos. Las causas pueden dividirse en 3 grandes grupos: Hipersecreción de insulina o péptidos relacionados con la misma, incremento en la utilización periférica de glucosa o falla en las hormonas contrarreguladoras. Caso clínico: Mujer de 37 años ingresa al Servicio de Clínica Médica por presentar hipoglucemias graves, hipertensión arterial e hipokalemia. Antecedentes personales: uso de medicación homeopática y trastornos de la conducta alimentaria. Menarca: 13 años, 1 embarazo, 1 parto. Amenorrea desde la suspensión del tratamiento con anticonceptivos orales por aumento de vello en mentón 3 meses antes. Examen físico: lúcida, ansiosa, adelgazada, TA: 180/100 mmHg, peso: 39kg, IMC: 16.25 Kg/m2, hemodinámicamente estable sin ortostatismo, score de Ferriman & Gallowey < 5. Laboratorio: Hepatograma y función renal: Normales, insulina: <1 uUI/ml (VR: 2-18), glucemia: 40mg/dl, Ks: 2 meq/l, FSH: <0.20 mUI/ml, LH: <0.20mUI/ml, E2: <20pg/ml, testosterona total: 7.8 ng/ml (0.90-1.3), 17OH progesterona: 49.40 ng/ml (<2), androstenediona 8.10 ng/ml (0.30-3.5), DHEAS: 9670 ng/ ml (VR: 350-4300), β-HCG negativo, IGF1: 40.2 ng/ml (VR: 95-285), IGFBP3: 3.9 mg/L (2-4), cortisol basal: 28.3 ug/ml, CLU: 102 ug/24hs (21-111), prueba de Nugent: 21.2l ug/d. 5-OH indolacético y catecolaminas urinarias, actividad de renina y aldosterona séricas: Normales. Ecografía transvaginal: Normal. TAC de abdomen y pelvis: lesión heterogénea de aspecto sólido de 13x12x12 cm sin plano de clivaje con respecto a parénquima hepático, intenso realce con contraste. Glándula suprarrenal izquierda normal, no se visualiza suprarrenal derecha debido a lesión anteriormente descripta. No adenomegalias. Se extirpa masa retroperitoneal, evoluciona sin complicaciones, normaliza valores de glucemia, presión arterial y kalemia. Tres meses después recupera ciclos menstruales y 8.5 kg de peso, IMC: 19.7 Kg/m2, LH: 4.6 mUI/ml, FSH: 5.1 mUI/ml, E2: 124 pmol/l, DHEAS 150 ng/ml, 17OH progesterona: 0.10 ng/ml, testosterona: 0.15ng/ml, testosterona biodisponible: 0.15 ng/ml (<0.15), androstenediona: 0.30 ng/ml, cortisol: 3.2 ug/dl, sodio: 136 meq/l, potasio: 4 meq/l, IGF-1: 63 ng/ml, IGFBP3: 0.4mg/L. PET-TC corporal con 18-FDG sin evidencia de tejido neoplásico. Anatomía patológica: Carcinoma adrenal (atipia citológica grado nuclear 3; compromiso capsular, componente de células claras menor de 25 % y necrosis). Actualmente recibe hidrocortisona 15 mg/día, pautas de stress. Continúa en seguimiento por servicio de Endocrinología y Oncología. Discusión: El carcinoma adrenocortical tiene una incidencia de 0.04 a 0.2 % entre todos los tumores malignos. La mayoría de las veces se manifiesta como tumor hiperfuncionante (más frecuentemente con hipercortisolismo y/o hiperandrogenismo) y rara vez con hipoglucemia. Los tumores NICTH (non-inslet cell tumor hyipoglycemia) se definen por la presencia de un tumor sólido con hipoglucemias en ayunas e insulinemia suprimida. La localización más frecuente es en tumores mensequimáticos (45 %), hepáticos (23 %), adrenales (10 %) y tubo digestivo (8 %). Se presenta este caso por la rareza del mismo. RAEM 2012. Vol 49 Nº Supl. 132 10 GINECOMASTIA: UNA FORMA INUSUAL DE PRESENTACIÓN DE LA HIPERPLASIA SUPRARRENAL CONGÉNITA (HSC) EN EL VARÓN ADULTO Pragier Um, Lutfi Rj. Sección Andrología , Servicio de Endocrinología del CMPFA Churruca Visca. Uspallata 3400 (C1437JCP), CABA, Argentina. upragier@hotmail.com, upragier@yahoo.com Introducción: La ginecomastia, crecimiento del parénquima mamario en el hombre, puede observarse en situaciones fisiológicas (ej: puberal) o patológicas (prevalecen los fármacos y causas idiopáticas). El denominador común es el disbalance estrógenos/testosterona. La HSC en el varón adulto puede manifestarse en el contexto del estudio por esterilidad (daño variable en espermatogénesis, hipogonadismo y/o alteraciones aisladas en gonadotrofinas o testosterona), a partir del hallazgo de restos adrenales testiculares (RAT) en una ecografía, o no diagnosticarse nunca. Objetivos: 1) Análisis de un caso de HSC que se presenta en el varón adulto con una ginecomastia, forma poco habitual, 2) Planteo de estrategias a seguir. Metodología: Varón de 26 años que consulta por aparición de molestias en mama izquierda, sobre una ginecomastia de larga data. Datos potencialmente relevantes: consumo esporádico de marihuana, ingesta de pollo 4 veces/semana. No consume medicación. Desarrollo puberal normal. Antecedentes maternos de cáncer de mama. Al examen físico, ginecomastia izquierda difusa (corroborado por mamografía). Aporta: 1) Hepatograma, función renal y tiroidea, CEA, alfa-fetoproteína y ß HCG normales. FSH 2 UI/L LH 5 UI/L prolactina 9 ng/ml testosterona 7,36 ng/ml testosterona biodisponible 13,98 (VR: 6,5-10,2 nmol/l), 2) Ecografía testicular normal. Se solicita 17OH progesterona: 7,5 mcg/l. Se reconfirman valores hormonales. Se indica prueba de ACTH. Resultados: 17OH progesterona basal: 3,8 60 minutos: 14,6. Cortisol basal 18,7 60 minutos 25. Se confirma diagnóstico de HSC. Se solicita estudio genético (CYP21) y espermograma. Se indica tamoxifeno 20 mg/d. Conclusiones: La descripción de este caso ilustra sobre la necesidad de recordar el diagnóstico de HSC en un varón adulto con ginecomastia, principalmente en presencia de alteraciones en las gonadotrofinas y/o testosterona. La ausencia de RAT no descarta posibles alteraciones, presentes o futuras, en el espermograma por lo que se recomienda la evaluación periódica del mismo y de la necesidad del uso de corticoides a tal fin, en cada caso. Bibliografía: 1. 2. 3. Gynaecomastia and breast cancer in men. Niewoehner C. et al. BMJ 2008:336(709-713). Testicular Tumors in Patients with Congenital adrenal Hyperplasia due to 21-Hydroxylase Deficiency Show Functional features of adrenocortical Tissue. Claahsen-van der Grinten H. et al. JCEM 2007:92 (3674-3680). Congenital adrenal hyperplasia in adults: a review of medical, surgical and psychological issues. Ogilvie C. et al. Clin Endoc. 2005:64(2-11). FASEN 2012 CASOS CLÍNICOS PRESENTACIÓN INFRECUENTE DE QUISTE SUPRARRENAL Mercado F, D´Jallad C, Torres I, Abate C, Maidana E, Cerioni V, Salvo MC, Gonza N, Galíndez M. Programa de Endocrinología, Hospital de Endocrinología y Metabolismo “Dr. Arturo Oñativia”. Eduardo Paz Chaín Nº 30. Salta CP 4400. Argentina. felicitasmercado@hotmail.com 133 11 Introducción: Los quistes suprarrenales son una entidad infrecuente cuya incidencia varía entre el 0,064 % y el 0,18 %. Generalmente son asintomáticos y su diagnóstico es incidental, sin embargo si son voluminosos pueden manifestarse por efecto de masa sobre las estructuras abdominales causando dolor abdominal, síntomas gastrointestinales o masa palpable en flanco. La fiebre y el dolor son síntomas derivados de la infección o hemorragia intraquística. Otros síntomas más infrecuentes son hipertensión arterial o ruptura espontánea del mismo con hemorragia retroperitoneal consecutiva. La mayoría son unilaterales y uniloculares. Hay predominio femenino y se presentan más comunmente entre la tercera y quinta década de la vida. Pueden medir desde milímetros hasta 50 cm de diámetro. Se clasifican en cuatro grupos: parasitarios (7 %), epiteliales (9 %), endoteliales (45 %) y pseudoquistes (39 %), siendo el pseudoquiste el subtipo más frecuente. A continuación se presenta un caso de presentación atípica de quiste suprarrenal. Caso Clínico: Paciente de sexo masculino que consulta por primera vez a los 20 años de edad. Refiere mareos, nauseas, cefaleas, ansiedad, sudoración, eritema facial y palpitaciones de presentación episódica desde los 4 años de edad, tratado como epilepsia con Carbamazepina hasta los 12 años cuando se suspende por intoxicación. Antecedentes heredo familiares de HTA en padre, muerte por IAM. Examen Físico: TA brazo izquierdo: 130/80 TA brazo derecho 120/70, FC 84 por minuto regular, resto normal. Estudios complementarios: Metanefrinas plasmáticas y urinarias, totales y fraccionadas normales. Ionograma plasmático normal, aldosterona normal, actividad renina plasmática normal. Cortisol plasmático matutino normal. TAC de abdomen: formación hipodensa de contornos definidos con pequeñas calcificaciones periféricas a nivel de glándula suprarrenal izquierda de 90 x 60 x 90 mm de tamaño mayor. Con diagnóstico de tumor suprarrenal no funcionante se interviene quirúrgicamente por vía laparoscópica. Durante la misma se evidencia tumoración quística de 8 a 9 cm de diámetro, evacuándose contenido. Se envía material a anatomía patológica que informa formación quística de 5 cm, multiloculada, de superficie lisa y en sectores rugosa con calcificación. Diagnóstico: quiste (pseudoquiste) adrenal, se sugiere realizar inmunomarcación, resultado pendiente. En el postoperatorio evoluciona con cuadro de similares características sin registro de HTA interpretado por psiquiatría como crisis de ansiedad con buena respuesta al tratamiento indicado con ansiolíticos. Conclusión: Los quistes suprarrenales son una patología poco frecuente y su diagnóstico suele ser incidental. El manejo del quiste suprarrenal se basa en tres pilares fundamentales: descartar estado funcional, evaluación de posible malignidad por imágenes y evitar sus complicaciones sobre todo en los quistes de gran tamaño. Se sugiere su abordaje laparoscópico excepto en lesiones de gran tamaño, malignidad o quistes parasitarios. El caso presentado anteriormente es de presentación atípica, tanto por la edad de presentación, el cuadro clínico sugerente de funcionalidad como la lesión multilocular. RAEM 2012. Vol 49 Nº Supl. 134 12 INSUFICIENCIA SUPRARRENAL SECUNDARIA A HISTOPLASMOSIS D’Jallad MC, De Los Ríos A, Gonza N, Galíndez M. Programa de Endocrinología, Hospital de Endocrinología y Metabolismo “Dr. Arturo Oñativia”. Dr. Eduardo Paz Chaín 30. Salta 4400, Argentina. macarenagalindez@hotmail.com Introducción: La insuficiencia suprarrenal es la consecuencia de la disminución de la síntesis de glucocorticoides por parte de la glándula suprarrenal asociada o no con la disminución de mineralocorticoides. Puede ser causada por enfermedades que comprometen a la glándula suprarrenal (Primaria), a la glándula hipófisis y en consecuencia a la secreción de ACTH (Secundaria) o que comprometen al hipotálamo y a la secreción de CRH (Terciaria). La etiología es variable, siendo las causas infecciosas las menos frecuentes. Caso clínico: Paciente de sexo masculino de 46 años, con diagnostico de DBT 2 en tratamiento que ingresa por náuseas y vómitos de 5 días de evolución y oscurecimiento progresivo de la piel. APP: Diabetes 2 de 12 años evolución con tratamiento desde hace 2 años (MTF e INS NPH 12 UI/día); amputación de 1er dedo de miembro inferior derecho; HTA (Enalapril 20 mg/día); Neuropatía DBT. Hipotiroidismo (Levotiroxina 50 mcg/día). Examen físico: Clínicamente estable con hiperpigmentación cutáneo generalizada, uniforme. Mucosas semihúmedas con máculas hiperpigmentadas. Pliegues en manos y pies hiperpigmentados, al igual que cara y cicatrices. Cuello: Tiroides OB. TA decúbito: 160/95 – bipedestación 140/85. Laboratorio: Cortisol 3,55(5-25 mcg/dl); TSH 4,15 ATPO (-); HbA1C 9,6 %; Na 139/K 3,63/Cl 104; FAN (-) ASMA (-) ANA (-); Prueba de estímulo con ACTH: basal 3,8; postest: 3,3. Hemocultivo (-); Urocultivo (-). TAC Abdomen: en Glándulas suprarrenales: formaciones expansivas sólidas bilaterales con contornos regulares y densidad de partes blandas. GSDer: 5 x 3 cm, GSIzq 5,5 x 3,4 cm. Dx: Lesiones neoformativas primarias o secundarias. Se trata con: Hidrocortisona. Es valorado por Infectología: Histoplasmosis (+) ¼ (inmunodifusión): Histoplasmosis diseminada subaguda. Se inicia tratamiento con Itraconazol 400 mg/día por 6 meses. CONTROL (6 meses): TAC abdomen de iguales características. Serologia Histoplasmosis (+) 1/4. Se decide continuar con igual tratamiento. Conclusión: La histoplasmosis, causada por el Histoplasma capsulatum, se adquiere por vía inhalatoria, afectando generalmente pulmones en el 90 % de los casos. Tiene diseminación hematógena en las primeras dos semanas, antes de que la inmunidad específica alcance a desarrollarse. Posteriormente la inmunidad celular controla la infección en pacientes inmunocompetentes lo que explica su curso subclínico y limitado. Menos del 5 % de los individuos expuestos desarrollan una enfermedad sintomática después de la exposición a bajo inóculo. La enfermedad de Addison por histoplasmosis es rara (10-20 %) aunque el compromiso suprarrenal es frecuente (80 %) en pacientes a los cuales se les realiza TAC o ecografía de abdomen. En nuestro caso se asume el diagnóstico de Insuficiencia Suprarrenal por Histoplasmosis esperando su confirmación por biopsia FASEN 2012 CASOS CLÍNICOS PROLONGADA REMISIÓN ESPONTÁNEA DE SÍNDROME DE CUSHING: ¿CURACIÓN O CICLICIDAD? Delfino L, Fuentes A, Pardes E. 135 13 División Endocrinología, Hospital J.M. Ramos Mejía, CABA, Argentina Introducción: La remisión espontánea (RE) del Síndrome de Cushing (SC) es un fenómeno infrecuente y mayormente vinculado a necrosis de tumor hipofisario. Existen referencias de RE prolongada en el contexto de un Cushing cíclico así como también otros autores llegan a definirla como una curación-semejante a la obtenida por adenomectomía. Presentamos una paciente con un cuadro clínico florido de Cushing de corta evolución, que inesperada y rápidamente entra en RE al mes de la consulta, persistiendo asintomática y eucortisolémica desde hace 2 años. Caso clínico: Paciente de 17 años derivada a Endocrinología por el Servicio de Neurocirugía presentando: debilidad muscular, aumento de peso, acné, estrías rojo vinosas y amenorrea 2° de 4 meses de evolución asociados a imagen de RM hipofisaria compatible con microadenoma de 7mm. La sintomatología coincidió con importante estrés familiar. Niega corticoides exógenos. Menarca: 12 años, eumenorreica hasta 4 meses atras. Examen físico: Peso: 78kg, TA 150/110mmHg (previamente normortensa), moderado acné facial y tóracoabdominal, acantosis nigricans en cuello, giba dorsal, almohadillas supraclaviculares, marcadas estrías rojo-vinosas >1cm en abdomen, muslos, axilas y región mamaria y disminución de masa muscular glútea. Impresión diagnóstica: SC, confirmado por (Ago´10): CLU 529,2µg/24hs, Nugent 19,8ug/dl, Cortisol salival 23hs 60nmol/l. Se confirma ACTH dependencia (ACTH 69,2pg/ml). Se solicita cortisol basal (18.9ug/dl) y post dexa-8mg (29,3 ug/dl) con respuesta paradojal. Se indica repetir. Se solicita TAC de tórax y abdomen (normales) y productos de co-secreción; se planifica cateterismo de seno petroso inferior. Por el cuadro rápidamente evolutivo, se indica tratamiento con Ketoconazol (KTZ) 400 mg/d, con controles seriados de hepatograma. A los 15 días de tratamiento: marcada elevación de transaminasas: se indica suspender el fármaco, Se solicita interconsulta con hepatología, que realiza los estudios correspondientes e interpreta el cuadro como hepatitis tóxica 2° a KTZ. A los 7 días de suspensión ,descienden las transaminasas. Se cita a la paciente cada 15 días, pero concurre luego de 2 meses: eumenorreica, facies afinada, no rubicunda, no giba, estrías en involución, fuerza muscular recuperada, normotensa (110/70mmHg) y ↓ peso (69,8kg). No refería cefaleas ni trastornos visuales Se solicita (Dic´10): Cortisol basal 5.3ug/dl CLU <12.2µg/24hs, ACTH 19,6pg/ml. (Ver Tabla 1). RNM hipofisaria (Ene´11): microadenoma de 5mm. La paciente se encuentra en control hasta la actualidad, presentándose clínicamente asintomática, eumenorreica, sin otros signos de disfunción hipotálamo-hipofisaria, hepatograma normal. Tabla 1. Datos Evolutivos de Laboratorio (2010-2012) Comentarios: 1. En nuestra paciente resulta llamativa la instalación del SC y su rápida evolución así como la brusca remisión espontánea que persiste a 2 años de su comienzo 2. Aunque en su RE no presentó cefaleas, podría igualmente tratarse de una remisión espontánea por necrosis focalizada, asintomática, que no fuera evidenciable por la RNM hipofisaria. 3. Se desconoce el rol que las citoquinas podrían desempeñar en la fisiopatología del cuadro (¿desencadenante?) 4. Hasta el momento, la paciente se encuentra sin estigmas de SC y eucortisolémica: consideramos que sólo el control a través del tiempo permitirá revelar si su RE corresponde a un período quiescente de un SC cíclico (aunque no tenemos evidencia de periodicidad) o un proceso de curación. RAEM 2012. Vol 49 Nº Supl. 136 14 FEOCROMOCITOMA ESPORÁDICO E HIPERCALCEMIA: PRESENTACIÓN DE UN CASO Villa S, Magat P, Vidal Armijo E, Bachini V, Costa L. Servicio de Endocrinología y Metabolismo Htal. Profesor A. Posadas, CABA, Argentina. Introducción: No es frecuente la asociación de hipercalcemia con feocromocitoma esporádico, existiendo pocos casos publicados en la literatura. El mecanismo fisiopatológico, en estos casos, puede ser explicado por el aumento de la secreción de PTH por acción directa de las catecolaminas o a la producción de PTHrp por el tumor. Presentamos un caso de feocromocitoma con hipertensión paroxística e hipercalcemia que se corrige luego de la adrenalectomía. Caso clínico: Varón de 40 años nacido en Bs As que consulta en Julio de 2010 por presentar diarrea secretoria de 8 meses de evolución asociada a dolor abdominal, náuseas, pérdida de peso de 10 Kg y trastornos conductuales (agresividad) 15 días previos. Se constata hipercalcemia, hipopotasemia e insuficiencia renal aguda prerrenal y en ecografía abdominal formación heterogénea mixta que se proyecta en región suprarrenal derecha de 75X93X67mm con área central de necrobiosis o degeneración quística y escasa vascularización en su interior (IR 0.49). Se interna para estudio de tumor suprarrenal derecho e hipercalcemia. Se indica plan amplio de hidratación y dieta astringente. Es evaluado por nuestro servicio: se rescatan antecedentes de un episodio aislado de HTA con cefalea y sudoración en el 2004 y litiasis renal. Examen físico paciente lúcido, orientado en tiempo y espacio, TA110/70 mmHg, FC 68X’, sin hiperpigmentación de piel y mucosas, tiroides palpable a la deglución y debilidad muscular generalizada. Lab. CaT mg/dl 12.3 Cai mmol/l 1.8 P Cau FAL mg/dl (2900cc) UI/l 3.7 513 147 PTH pg/ml 8 25(OH)D3 ng/ml 12.6 Alb g/l Iono mmol/l glu g/l 4.1 135/2.6/105 128 urea g/l Cr mg/dl 0.84 1.5 Adrenalina U ug/24hrs Noradrenalina ug/24 hrs AVM mg/24 hrs Cortisol pl ug/dl T de Nugent ug/d CLU ug/24 hrs 100 VN hasta 27 470 VN hasta 97 11.8 VN hasta13.6 21.3 VN 8.7-22.4 0.8 103.8 (VN 21-143) Eco tiroidea normal. TSH 0.11uUI/ml, T4L 1.01 ug/dl, ATPO 0.2 U. TAC Abd/pelvis sin cte: lesión primaria de glándula suprarrenal derecha de 7x7cm. RNM con gadolinio: Formación nodular heterogénea que compromete glándula suprarrenal derecha con realce periférico heterogéneo a nivel de la cápsula. Suprarrenal izquierda s/p. Se solicita centellograma óseo: sin evidencia de zonas patológicas. El proteinograma electroforético fue normal. Se inició sustitución con Vit D3. Por persistencia de hipercalcemia moderada a pesar de la hidratación, se indicó Pamidronato 30 mgr/ev. Durante la internación intercurre con crisis hipertensiva (TA170/100) asociado a sudoración profusa y cefalea. Con diagnóstico clínico y bioquímico de feocromocitoma se inicia tratamiento con alfa bloqueantes y se decide resolución quirúrgica. Se realiza suprarrenalectomía derecha laparoscópica, intercurriendo en el intraoperatorio con hipertensión severa, requiriendo tratamiento con nitroprusiato de sodio con buena respuesta. En el postoperatorio, evoluciona normotenso, sin diarrea, normocalcémico, normopotasémico. Se recibe resultado de anatomía patológica que confirma el diagnóstico de feocromocitoma. Fenotipo: Vimentina (+) 20 %, (3+) células neoplásicas, CK (-), CK7 (-), CK20(-), Cromogranina (+) 80 % (3+) células neoplásicas, Sinaptofisina (+) 80 % (3+) células neoplásicas, CD 56 (+) 80 % (3+) células neoplásicas, TTF1 (-), Ki-67 (+) 1%, MELAN A (-). Lab post quirúrgico: CaT corregido por albúmina 9.48 mg/dl; Mg 1.9 mg/dl; P 5,5 mg/dl; iono 141/4.9/112 mmol/L, glu 0.80 g/l, u 0.25g/l; cr 1.2mg/dl. Conclusión: La asociación de hipercalcemia y feocromocitoma generalmente se observa en el MEN II. En nuestro paciente, los valores de PTH plasmáticos bajos en relación al nivel de calcemia, y la correción de la hipercalcemia luego de la cirugía sugiere fuertemente la producción y secreción de PTHrp por el tumor. FASEN 2012 CASOS CLÍNICOS SÍNDROME DE CUSHING ACTH-DEPENDIENTE, CORTICOTROPINOMA Y EMBARAZO Juárez-Allen L, Urciuoli CC, Politi S, Gómez RM, Herrera JD, Bruno OD. Hospital de Clínicas, UBA, Argentina. 137 15 Introducción: El embarazo no es habitual en pacientes con síndrome de Cushing (SC), ya que el exceso de cortisol y eventualmente de andrógenos, interfiere con la secreción de gonadotrofinas así como también con el desarrollo folicular normal y la ovulación. El seguimiento y tratamiento son controvertidos. Existen alrededor de 150 casos publicados en la literatura mundial. Objetivo: Describir la evolución y manejo del embarazo en 5 pacientes con diagnóstico previo de SC ACTHdependiente (SC-A). Pacientes: Entre 1998 y 2011, 5 pacientes con SC-A lograron embarazos (ver tabla). La paciente nº 1 se embarazó en el curso de su evaluación y consultó tras 6 meses de amenorrea, sin conocimiento de su condición. Las pacientes 2 a 4 tenían enfermedad de Cushing con corticotropinomas probados por patología en tanto que la paciente 5 presentaba un síndrome de Nelson con un corticotropinoma evidente en la resonancia magnética nuclear. Todas lograron embarazos de manera espontánea, excepto la paciente 5, que lo logró mediante fertilización asistida (FIV). Paciente 1 Paciente 2 Paciente 3 Paciente 4 Paciente 5 Edad al diagnostico 27 21 22 26 24 Edad al embarazo 30 32 25 28 35 x FIV Tratamiento pre- embarazo -- TSE CBG KTZ TSE KTZ TSE 1°TSE 2°TSE ADX bilateral γ-Knife Recidiva/ persistencia -- SI - SI SI Síndrome de Nelson CLU 24 pre embarazo (µg/24hs) 215 254 198.2 97.2 86.0 CLU 24 en el embarazo* (µg/24hs) 96 496 1372 274 198 Tratamiento durante embarazo NO NO KTZ NO (2° trimestre) por HTA / DBT Hidrocortisona Betametasona Evolución fetal Muerte fetal semana 33 RNT Sano RNPT Sano RNT Sano RNT Sano TSE: Cirugía transesfenoidal, ADX: adrenalectomía, CLU cortisol libre urinario (vn 20-90),CBG: Cabergolina, KTZ: Ketoconazol, HTA: Hipertensión arterial, DBT: Diabetes, RNT: Recién nacido a término; RNPT: Recién nacido pretérmino . * valor más elevado observado. Discusión: El SC durante el embarazo está asociado con severas complicaciones maternas, HTA, DBT, preeclampsia e insuficiencia cardiaca y fetal, como parto prematuro, aborto espontáneo, mortalidad intrauterina, retardo de crecimiento intrauterino e insuficiencia suprarrenal. Su diagnóstico y tratamiento temprano es fundamental para lograr un embarazo a término. La decisión de realizar tratamiento durante el embarazo y su elección dependen de la etiología, la gravedad de la enfermedad y la edad gestacional. El mismo debe ser individualizado en cada caso aunque se ha sugerido que el tratamiento de elección es la cirugía al final del 1er trimestre o durante la primera mitad del 2do. Otras alternativas incluyen adoptar una conducta expectante, tratando las complicaciones que se presenten o indicar un tratamiento farmacológico, siendo la droga de elección la metopirona. Conclusión: Pese a que el embarazo en pacientes con SC no tratado se asocia con una elevada morbilidad materna y fetal, en nuestra experiencia la observación y el seguimiento cuidadoso de las pacientes nos permitió mantener una conducta expectante que, en algunos casos, resultó en embarazos sin complicaciones y recién nacidos sanos. RAEM 2012. Vol 49 Nº Supl. 138 16 MIOCARDIOPATÍA TIPO TAKO TSUBO POSTRAUMÁTICO COMO FORMA DE PRESENTACIÓN DE FEOCROMOCITOMA Bustamante MA1, Deutsch SI1, Szelubsky S1, Botvinik, G2, Wolanow, V3, Calvar Ce1 1 Servicio de Endocrinología, 2Servicio de Cardiología, Sección Hipertensión Arterial, 3Unidad de Terapia Intensiva; Hospital General de Agudos “J. A. Fernández”, Cerviño 3356 (1425), CABA, Argentina. endocrinofernandez@yahoo.com.ar Tel. 4808-2600 Objetivo: Reportar el caso de una paciente con feocromocitoma cuya manifestación inicial fue una miocardiopatía tipo tako-tsubo. Reporte del caso: Paciente sexo femenino de 46 años, sin antecedentes patológicos, ingresó a Emergentología en shock hipovolémico secundario a trauma lumbar. La ecografía abdominal mostró líquido libre en cavidad abdominal e imagen compatible con laceración esplénica. Se realizó laparotomía con esplenectomía. Durante la cirugía se evidenció una masa retroperitoneal izquierda, no pulsátil, no expansiva. En el postquirúrgico inmediato presentó dolor precordial típico con cambios en el electrocardiograma (supradesnivel del ST de V1 a V6) y elevación de enzimas cardíacas. Se interpretó como miocardiopatía tipo tako-tsubo por la presencia de hipoquinesia global y aquinesia del ventrículo izquierdo en ecocardiograma y coronariografía sin evidencia de obstrucción de arterias coronarias. Ingresó a Terapia Intensiva requiriendo ventilación mecánica por 4 días, APACHE II de 27 puntos y probabilidad de mortalidad 25 %. La tomografía abdominal postquirúrgica evidenció formación sólida heterogénea redondeada de 85x75mm en topografía de glándula suprarrenal izquierda. Catecolaminas urinarias: Adrenalina: 39.9 ug/24hs (VR 0-8.5); Noradrenalina: 477 ug/24hs (VR 18.5-100); Àcido Vainillin Mandélico: 28 ug/24hs (VR 1.8-8.5) En agosto de 2011 inició tratamiento con carvedilol y doxazosina y en septiembre del mismo año se realizó resección de glándula suprarrenal izquierda. Anatomía patológica: feocromocitoma, tamaño tumoral de 8x7.5x4, 5 cm, presencia de necrosis y hemorragia intratumoral, infiltración de cápsula tumoral y glándula suprarrenal adyacente, inmunomarcación positiva para sinaptofisina, enolasa NE y Ki-67: 6 %. Tras la cirugía la paciente presentó hipotensión arterial que respondió a inotrópicos. Actualmente se encuentra asintomática con valores de catecolaminas urinarias normales y ecocardiograma normal. Discusión: La presentación clínica más frecuente de los feocromocitomas es un síndrome caracterizado por hipertensión arterial severa, acompañado de cefalea, taquicardia y diaforesis. Las manifestaciones cardíacas incluyen arritmias, anormalidades en el electrocardiograma, hipertrofia ventricular, síndromes coronarios agudos, falla cardíaca congestiva secundaria a miocarditis o miocardiopatía y shock cardiogénico. La miocardiopatía tipo tako-tsubo es un síndrome de aturdimiento miocárdico intenso, precipitado por situaciones de estrés agudo, más frecuentemente descripto en mujeres. Se ha demostrado que la liberación suprafisiológica de catecolaminas provenientes de un feocromocitoma, disminuye la viabilidad de los miocitos a través de una sobrecarga de calcio mediada por AMP cíclico y producción de radicales libres que sería la responsable de la cardiotoxicidad. Se caracteriza por disfunción ventricular izquierda severa, reversible, transitoria que produce abalonamiento apical con hiperkinesia basal compensatoria, cambios en electrocardiograma y elevación de enzimas cardíacas simulando un infarto agudo de miocardio pero sin enfermedad coronaria obstructiva por cinecoronariografía. Si bien la miocardiopatía tipo tako tsubo es una forma poco frecuente de presentación de una crisis adrenérgica, debe tenerse en cuenta en el diagnóstico diferencial de un síndrome coronario agudo por feocromocitoma. En nuestra paciente el traumatismo lumbar y el procedimiento quirúrgico precipitaron este tipo de miocardiopatía, que sumado a la visualización de la masa adrenal llevó al diagnóstico de feocromocitoma. FASEN 2012 Viernes 5 de octubre CASOS CLÍNICOS - Gónada masculina, Hipófisis y Varios - 17 al AGRANDAMIENTO TESTICULAR EN PACIENTE CON GONADOTROPINOMA PRODUCTOR DE FSH 1 1 139 24 - Salón Castilla + Villalba - Coordinador: H Boquete. Mana D , Belingeri MS , Lopardi M , Miño A , Peirano M , Manavela M , Cazado E 1 2 3 4 1 17 Sección Endocrinología 2Laboratorio y 3Sección neurocirugía del Hospital de Agudos J.M. Penna. 4División Endocrinología Hospital de Clínicas J. de San Martín Pedro Chutro 3380, 1437 CABA, Argentina. daniela.mana@gmail.com 1 Introducción: Se conoce que el 80-90 % de los adenomas hipofisarios clínicamente no funcionantes son gonadotropinomas. La mayoría, sin embargo, secretan bajas concentraciones de FSH, LH o sub-unidades. Endocrinológicamente son silentes, presentándose solo con síntomas de efecto de masa o alteraciones visuales. Gonadotropinomas con altas concentraciones de FSH o LH, y con signos y síntomas de hipersecreción de dichas hormonas han sido raramente reportados. Caso clínico: Paciente varón de 51 años de edad con cuadro de 10 años de evolución caracterizado por alteración de la conducta y del equilibrio, depresión e incontinencia de esfínteres. Hace 1 año agregó alteraciones visuales. Ingresó a nuestro hospital por presentar traumatismo encefalocraneano con pérdida de conocimiento en el contexto de episodio convulsivo. La tomografía computada mostró una extensa formación tumoral selar y supraselar con destrucción de ambas apófisis clinoides. Al examen físico se encontró amaurosis del ojo derecho con visión bulto del ojo izquierdo y signos de hipogonadismo, con un llamativo aumento del volumen testicular bilateral (mayor a 25 ml). La resonancia nuclear magnética mostró en proyección selar y supraselar voluminosa lesión que ejerce efecto de masa sobre el tronco cerebral y se proyecta hacia el piso del 3º ventrículo. Refuerza en forma homogénea luego del contraste. En la valoración hormonal presentaba FSH 517 mUI/ml (Valor Normal-VN 1.5-12.4), LH 2.2 mUI/ml (VN 1.78.6), Testosterona total 4.47 nmol/L(VN 9.9-27.8), Prolactina 21.2ng/ml (VN 4.6 -21.4), TSH 1.37 µUI/ml(VN 0.274.2), T4L 0.77 ng/dl(VN 0.9-1.7), Cortisol matinal 4.2 µg/dl (VN 6.2-19.4), IGF1 33 ng/ml(VN 87-200), Inhibina B: 236 pg/ml (VN 230±42) y HAM:5.3 (VN 3-8) Se realizó un primer procedimiento quirúrgico (previo reemplazo cortical y tiroideo) por vía fronto pterional con extracción del 30% del tumor y se re-operó por igual vía a los 12 días con mayor resección tumoral. La anatomía patológica informó proliferación neoplásica, dispuesta en nidos y en un patrón papilar, constituida por células con citoplasma amplio anfófilo y en otros eosinófilos. Inmunohistoquímica: marcación heterogénea y amplia para FSH, en alto porcentaje (cercano al 70 %); y negatividad para ACTH, Prolactina, LH y TSH. El índice de proliferación con Ki-67 fue menor a 1 %. Diagnóstico: GONADOTROPINOMA PRODUCTOR DE FSH. La valoración hormonal posquirúrgica mostró una caída de los valores de FSH a 22.35 mUI/ml. El paciente presentó evolución tórpida. Requirió colocación de derivación ventrículo peritoneal por hidrocefalia con múltiples complicaciones infectológicas, requiriendo asistencia ventilatoria mecánica y sostén hemodinámico, con posterior óbito. Discusión y conclusión: En el hombre la FSH estimula las células de Sértoli y el túbulo seminífero ejerciendo un importante efecto trófico testicular favoreciendo la espermatogénesis. Esto explicaría el gran tamaño testicular presentado por nuestro paciente en respuesta a las altas concentraciones de FSH. Dichos valores son los más altos reportados en la literatura hasta la actualidad para gonadotropinomas secretores de FSH. Inicialmente la LH se encontraba en rango inferior bajo y la testosterona subnormal, lo cual se interpretó en el contexto de hipopituitarismo secundario al efecto de masa producido por el macroadenoma. Este caso enfatiza la importancia del examen testicular en pacientes hipogonádicos ya que es un signo clínico fundamental que podría hacernos sospechar la presencia de un adenoma hipofisario productor de gonadotrofinas. RAEM 2012. Vol 49 Nº Supl. 140 18 PACIENTE CON SÍNDROME DE EUNUCO FÉRTIL (SEF): RESOLUCIÓN DE INFERTILIDAD PRIMARIA Balonga MC, Politi S, Cazado E, A. de Cross G. Hospital de Clínicas. UBA. CABA, Argentina Introducción: El factor masculino como causa de infertilidad se observa en el 30 % de los casos, tanto en forma aislada como asociado a factor femenino. El SEF describe al paciente hipogonádico eunucoide con volumen testicular normal y es causa infrecuente de infertilidad primaria. Su etiopatogenia y tratamiento es aún motivo de discusión y controversia. Caso clínico: Un paciente de 34 años consulta por infertilidad, luego de haber descartado factor femenino. Al examen físico presenta hábito eunucoide (braza 198 cm, talla 188 cm, segmento superior 95 cm, segmento inferior 93 cm) distribución ginecoide de la grasa corporal y ausencia de caracteres sexuales secundarios masculinos, vello pubiano Tanner 4, volumen testicular izquierdo y derecho 18 cc cada uno, sin engrosamiento del cordón espermático y pene 6 cm. La función sexual se encuentra conservada. Niega parotiditis, traumatismos testiculares, uso de drogas ilícitas y tabaco. Ante la presunción diagnóstica de hipogonadismo se solicita laboratorio y espermograma (Tabla 1 y 2). El Doppler testicular, el cariotipo y la RNM selar fueron normales. La olfatometría informó hiposmia leve. Tabla 1. Laboratorio 12/09 03/11 12/11 LH ( 0.8-7.6 mU/ml) 04/12 0.8 FSH (0.7-11.1 mU/ml) 1.4 Tº (2.6-10.12 ng/ml) 0.77 TºBD (1.2-6.3 ng/ml) 0.18 PRL (2.5-17 ng/ml) 10.3 TSH (0.4-4 uU/ml) 2.7 E2 (h/56 pg/ml) <20 TRATAMIENTO 6.9 2.4 Tabla 2. Espermogramas 3.3 06/09 03/11 Vol. (1.5-7 ml) 0.2 0.8 1.2 2.3 Grado A+B (>=50%) 3 14 17.8 29.3 Necrosados (<50%) 82 60 50 50 Zoides/ ml (>20x10 6) 10x106 66x106 Zoides/ eyac (>4 x10 6) 2x10 6 hCG Testosterona Testosterona Enantato Enantato Zoides Normales (>15%) 8 8/11 04/12 26x106 25x106 52.6x10 6 3 x106 58.8x106 5 0 9 Tº: Testosterona total. TºBD: testosterona Biodisponible. E2: Estradiol. hCG:Godadotrofina Coriónica Humana. Con la sospecha de SEF se inicia tratamiento de estimulación hormonal con hCG 2500 UI/bisemanal durante 6 meses, incrementándose luego a 2500 UI/trisemanal hasta completar 9 meses, continuando con la dosis inicial por 6 meses más. En octubre de 2011 informa embarazo de 7 semanas, por lo que se rota esquema a testosterona enantato IM cada 28 días. La gestación transcurre sin complicaciones, con RN varón sano. Discusión: Los pacientes con SEF, cuadro descripto por primera vez en 1949 por los Doctores Pascualini y Bur, presentan un “aparente” déficit parcial de gonadotrofinas (FSH y LH) lo cual resulta suficiente para obtener concentraciones de testosterona intratesticular adecuada para iniciar la espermatogénesis y, por consiguiente, aumentar el volumen testicular pero insuficiente para lograr el desarrollo de caracteres sexuales secundarios. La etiopatogenia es incierta. Inicialmente se incluía dentro del grupo heterogéneo de pacientes con Hipogonadismo Hipogonadotrófico Idiopático (HHI). En 2001 se describió una mutación homocigota (Gln 106 Arg) en el GnRHR en un paciente de 26 años que generaba baja afinidad de unión entre la hormona y su receptor. Este reporte provee la primera base genética en el SEF que demuestra un defecto parcial inactivante del GnRHR. Hasta el momento no se han descripto otras mutaciones causales del síndrome. El tratamiento consiste en el remplazo hormonal, inicialmente con hCG hasta lograr un adecuado volumen testicular y virilización completa para continuar, en lo casos no reversibles, con ésteres de testosterona IM o dérmica. Conclusiones: Frente a un paciente con hipogonadismo hipogonadotrófico y volumen testicular adulto debe evocarse el diagnóstico de SEF. El tratamiento oportuno con hCG sería apropiado para conseguir la virilización adecuada y asegurar la fertilidad futura. Los avances en biología molecular permitirán conocer la etiopatogenia de éste síndrome, permitiendo el asesoramiento genético y el tratamiento y seguimiento adecuado de la descendencia. FASEN 2012 CASOS CLÍNICOS HIPOGONADISMO HIPOGONADOTRÓFICO AISLADO: PRESENTACIÓN DE UN CASO Seleme S, Giacoia E, Díaz Sotelo P, Sciorra JM. Servicio de Endocrinología y Metabolismo, Hospital Nacional Prof. Dr. Alejandro Posadas. CABA, Argentina. 141 19 Introducción: El Hipogonadismo Hipogonadotrófico (HH) se caracteriza por una falla en la síntesis o secreción de la hormona liberadora de gonadotrofinas (GnRH) o de las gonadotrofinas (FSH/LH). Puede ser un déficit selectivo o asociado a otras hormonas. Se describen causas congénitas (con y sin anosmia), como el Síndrome de Kallmann, y defectos genéticos en la pulsatilidad del GnRH o adquiridas como adenomas hipofisarios, enfermedades granulomatosas, traumatismos, hemocromatosis, autoinmunidad o idiopáticas entre otras. El HH idiopático con presentación en la edad adulta es muy poco frecuente. Nosotros presentamos un caso de un varón que a los 35 años desarrolla manifestaciones clínicas y bioquímicas compatibles con HH luego de un desarrollo puberal y adultez normal. Caso clínico: Nuestro paciente consulta un año después del inicio de los síntomas por disminución de la libido y disfunción sexual eréctil, asociado a disminución de la fuerza muscular y menor crecimiento de barba. Antecedentes personales: criptorquidea bilateral con resolución quirúrgica a los 6 meses de vida. Dislipemia. Niega consumo de alcohol, tabaquismo y drogas de abuso. Antecedentes familiares: dislipemia madre y padre. Examen físico: peso: 75 kg, talla: 173 cm (IMC 25), sentido del olfato conservado sin ginecomastia ni galactorrea; disminución del vello pubiano, despigmentación del escroto y ambos testículos de 20 ml, estimado por el orquidómetro de Prader. Laboratorio: FSH 0.8mUI/ml, LH 0.2mUI/ml, Prolactina 6.6ng/ml, Estradiol <40pg/ ml, Testosterona total 0.24ng/ml, Cortisol 18.4ug/dl, TSH 2.53uUI/ml, T4l 0.95ng/dl, T4 9.9ug/dl, IGF1 179 ng/ ml, GH 0.17 ng/ml, ACTH 13.9 pg/ml, Ferritina 267 ng/ml, Colesterol total 276, HDL 48, LDL 181, TG 231. Se realiza prueba de estimulo con HCG: testosterona basal 0.20 ng/ml y post HCG 72 hs de 2.42 ng/ml. ATPO neg, Anti músculo liso neg, Ac antinucléolo neg, anti ADN neg, anti adrenal neg. No se realiza prueba LH/RH por falta de disponibilidad en el mercado. RMN de área selar con y sin gadolinio dentro de parámetros normales. Espermograma: Vol 0.4 ml, color blanco ligeramente opalescente, PH 8, licuefacción 60 minutos, viscosidad normal, no se observan espermatozoides, leucocitos 0.20 por 10 6 /ml. Fructosa 19.4 µmol/eyac, Acido cítrico 17.2 µmol/eyac. Con sospecha de Hipogonadismo Hipogonadotrófico aislado, se inicia tratamiento con Testosterona IM. Control a los 10 días: Testosterona Total 5.15ng/ml. El paciente refiere aumento de la fuerza muscular, con mayor crecimiento de barba. Actualmente continúa en tratamiento con Testosterona intramuscular, sin deseo de reproducción. Conclusión: Frente al resultado de los estudios realizados y a la adecuada respuesta al tratamiento instituido, que revirtió la mayoría de los síntomas de nuestro paciente, nos permitimos pensar que estamos frente a un cuadro de Hipogonadismo Hipogonadotrófico Aislado. RAEM 2012. Vol 49 Nº Supl. 142 20 METÁSTASIS GANGLIONAR DE CARCINOMA TALL CELL CON PRIMARIO OCULTO. REPORTE DE UN CASO Chiesa LF*, Attanasio RA*, Vera NV*, Menvielle S**, Cozzi S*** *Servicio de Endocrinología; **Servicio de Cirugía; ***Servicio de Patología; Hospital Profesor Dr. Rodolfo Rossi, La Plata, Buenos Aires, Argentina. Paciente masculino de 58 años oriundo de La Plata, derivado al Servicio por presentar tumoración latero cervical derecha supraclavicular. Antecedentes personales y familiares no relevantes. Enfermedad Actual: comienza dos meses previos a la consulta con una masa pequeña (1 cm aprox.) que descubre el paciente y que aumenta de tamaño rápidamente hasta alcanzar a la palpación 4x8 cm. Examen Físico: masa dura, consistencia pétrea, móvil respecto de planos profundos y piel, borde regular, no dolorosa en región supraclavicular derecha (Sector V). Se palpa Tiroides discretamente aumentada de tamaño (35 gr). No se palpan adenomegalias en el resto del cuello. Resto del examen sin particularidades. Se realiza Ecografía de cuello que informa: tiroides normal, masa lateral cervical derecha de aspecto sólido, heterogénea, de 5.4x3.8 cm. TAC de cuello y macizo facial: adenomegalias latero cervicales derechas entre 1 y 2 cm con una dominante en hueco supraclavicular de 5 cm. PAAF: cuadro citológico compatible con Carcinoma papilar de Tiroides de células altas (TALL CELL). Se realiza tiroidectomía total y vaciamiento cervical derecho. Del estudio anatomo-patológico se desprende: 1) Tiroides: cuadro histológico compatible con tiroiditis, ausencia de carcinoma (cortes de 3 mm) 2) Grupos ganglionares sectores II. III. IV. Y VI. Sin signos de infiltración por células atípicas. Del grupo ganglionar V se disecan seis ganglios sin signos de infiltración por células atípicas, observándose dentro del parénquima fibroadiposo, proliferación de células atípicas compatible con Carcinoma papilar de tiroides variedad clásico y de células altas. Se realizó marcación con tiroglobulina que fue positiva. El paciente recibió Dosis Ablativa con I 131 (150 mCi) con captación negativa. Nuevo rastreo a los 8 meses fue negativo y la tiroglobulina liberada fue < de 0,2ng/ml Conclusiones: Los tumores Tall Cell son una variante del carcinoma papilar de tiroides; de conducta más agresiva, mayor incidencia de recurrencia (36% al 58%) y mortalidad (18 % al 25 %). Representan el 7% al 12% de los carcinomas papilares. Si bien su forma de presentación por lo general es: en pacientes de edad avanzada (media 55 años) con invasión local y metástasis regionales, lo que implica alto riesgo, hay un riesgo elevado inherente al tipo histológico. Muchos muestran refractariedad al tratamiento con I131 y la PAAF no es un método eficaz para diagnóstico preoperatorio. Pueden tener metástasis a distancia sin enfermedad local avanzada o como primera manifestación de recurrencia. Destacamos este caso por el comportamiento no habitual de presentación ya que: 1. No se halló tumor en tejido tiroideo. 2. La localización de las metástasis fue en sector V habiendo salteado las principales estaciones ganglionares previas. 3. En el estudio histopatológico no se distingue tejido ganglionar invadido hallándose el carcinoma sobre tejido fibroadiposo lo que supone disrupción de la cápsula ganglionar. 4. Fue diagnosticado por PAAF preoperatoria. 5. Siendo un carcinoma papilar, no se halló captación en el rastreo post- dosis ablativa. La hipótesis de carcinoma sobre tiroides ectópica lateral fue desechada por su ubicación ya que estos restos tiroideos se localizan medial a la vaina carotidea. Bibliografía - Monteros M; et. al. Microcarcinomas papilares de Tiroides No Incidentales. Medicina (Buenos Aires) 2008; 68: 139-143 - Watkinson John C; et al. Detection and Surgical Treatment of Cervical Lymph Nodes in Differentiated Thyroid Cancer. Thyroid. Volumen 16, Number 2, 2006. - Angel Andres y col. Carcinoma Papilar de Tiroides, Variante de Células Altas. Rev. CES Medicina. 1999 Vol. 13 Nº 1:75-79 FASEN 2012 CASOS CLÍNICOS NEM 2A. IMPORTANCIA DEL SCREENING GENÉTICO EN LA TOMA DE DECISIONES Gonza N, Galíndez M, Van Cauwlaert L, Monteros Alvi M. Programa de Endocrinología, Programa de Cirugía, Sector Anatomía Patológica, Hospital de Endocrinología y Metabolismo Dr. Arturo Oñativia. Salta 4400. Argentina. dranataliagonza@gmail.com 143 21 Introducción: La Neoplasia Endócrina Múltiple tipo 2 constituye una referencia en el manejo del paciente con cáncer relacionado a la Genética. La NEM 2A es la más frecuente de este grupo de síndromes hereditarios, se caracteriza por presentar de manera sincrónica o metacrónica: Cáncer Medular de Tiroides, Hiperparatiroidismo y Feocromocitoma. Existen 2 variantes: la Amiloidosis Cutánea Liquenoide y la Enfermedad de Hirschprung . Se transmite de forma autosómica dominante, con penetrancia casi completa y de expresión variable. Se debe a mutaciones germinales del proto-oncogen RET, localizado en el brazo largo del cromosoma 10 banda 11 q2, que codifica para un receptor de la familia de las tirosina-kinasas. El CMT tiene una alta penetrancia y se presenta en casi el 100 % de los pacientes. Por lo general es el primer tumor en manifestarse clínicamente de forma bilateral y multicéntrica. La pesquisa del RET debe realizarse en todo paciente con CMT para discriminar entre la forma Esporádica y la Hereditaria. Entre el 1-7 % de los casos aparentemente esporádicos tienen mutaciones germinales RET, incluyendo 2-9 % de mutaciones “de novo”. La detección temprana de los familiares portadores de la mutación RET evita el desarrollo del CMT realizando una tiroidectomía profiláctica y obliga a descartar la expresión de otras endocrinopatías. Caso clínico: Mujer de 29 años hipertensa citada para pesquisa oncológica por madre portadora de cáncer medular de tiroides. Ecografía tiroidea: nódulos sólidos hipoecogénicos con microcalcificaciones vascularización tipo III de 14, 13, 8 y 16 mm der. e izq. PAAF: de nódulos en LI y LD Carcinoma Medular de tiroides, BETHESDA: 6. Calcitonina en lavado de aguja >2000 pg/ml. Eutiroidea, TPO:12,4 UI/ml,calcitonina:108pg/ml, CEA:44,2ng/ml, PTH: 131pg/ml, Ca:9,7mg/dl, Alb:4,7gr %, P:2,7mg %, Mg:2,2mg %, Cau24hs:512, Normetanefrinas O 24hs:317ug, Metanefrinas u 24hs:79ug. SESTAMIBI PARATIROIDEO: Imagen hipercaptante a nivel del polo tiroideo inferior izquierdo que persiste en las tomas tardías. Se realiza tiroidectomía total, vaciamiento central y paratiroidectomía inferior izq. A.P: carcinoma medular 14 mm intratiroideo multicéntrico asociado a hiperplasia de células C nodular y focal. Hiperplasia paratiroidea pseudoadenomatosa. MTS en 2/2 ganglios centrales y 3/6 recurrenciales izquierdos. T1N1Mx .Estadio III. ESTUDIO MOLECULAR: codón 634 del exón 11 del ret por PCR/RFLP sin alteración. Se repite el estudio: la secuenciación directa del exón 15 mostró una variación en la secuencia Val871Ile, GTT-ATT, Valina-Isoleucina compatible con cáncer medular de tiroides familiar. Se detectaron dos polimorfismos uno en el intrón 14, IVS14- 24G A y otro en el exón 15 Ser904. Ser heterocigota del proto oncogén RET. Evoluciona HTA. Adrenalina urinaria: 160 ug/24hs, noradrenalina urinaria 2225 ug/24hs. Se realiza adrenalectomía total izquierda y subtotal derecha. A.P: Feocromocitoma bilateral. Cromogranina (+) en células tumorales y proteína S 100 (+) en células subtentaculares. Ki 67 bajo índice de proliferación menor al 10 %. CEA, calcitonina, CK coctel: negativos. Evoluciona normotensa sin medicación antihipertensiva, con Hidrocortisona y Levotiroxina en dosis de reemplazo. Hija con enfermedad de Hirchprung en estudio para tiroidectomía profiláctica. Discusión: La coexistencia de CMT e hiperparatiroidismo y el antec familiar, orientan hacia el NEM 2A. En esta paciente, el estudio del proto-oncogen RET no detectó alteraciones en el codón 634 (mutación más frecuente). Se repite el estudio molecular identificándose dos polimorfismos del proto oncogén RET, no descriptos en la bibliografía, por lo que la sospecha clínica no debe desestimarse. RAEM 2012. Vol 49 Nº Supl. 144 22 XANTOMAS ERUPTIVOS: A PROPÓSITO DE UN CASO Eugenio Russmann M, Lusardi F, Portunato G, Virga M, Chiocconi M, Gauna A. Servicio de Endocrinología Hospital Ramos Mejía, CABA, Argentina. Introducción: Los xantomas, lesiones cutáneas formadas por macrófagos ricos en colesterol y triglicéridos alojados en el tejido celular subcutáneo, pueden constituir una manifestación clínica de alteraciones del metabolismo lipidico. Los xantomas eruptivos se asocian a dislipemias primarias o secundarias. Las dislipemias primarias tipo IV (Hipertrigliceridemia Familiar) y tipo V (hiperquilomicronemia familiar) son las que más se asocian con Xantomas eruptivos. La hipertrigliceridemia familiar, en la que se produce un aumento de las VLDL, tiene una prevalencia de 1/600 y se manifiesta como hepatoesplenomegalia, lipemia retinalis y xantomas eruptivos. La hiperquilomicronemia familiar, que se produce por un déficit de lipoproteinlipasa o de APO C II, presenta una clínica similar y se asocia a mayor incidencia de pancreatitis. La disbetalipoproteinemia, presenta una prevalencia de 1/10000 y se caracteriza por presentar xantomas palmares y tuberoeruptivos, cursa con aumento de triglicéridos y colesterol total y se asocia a mayor riesgo coronario. La DM es una de las causas secundarias más frecuentes de dislipidemia asociada con niveles elevados de triglicéridos, principalmente en DM1.Esto se debe a que la insulina mediante el estimulo de la lipoproteinlipasa regula el metabolismo de los quilomicrones y las VLDL, resultando en un aumento de los mismos en caso de insulinopenia o diabetes no compensada. Es importante reconocer hallazgos cutáneos que en un paciente asintomático pueden ser expresión de enfermedades sistémicas como la hipertrigliceridemia severa o la diabetes mellitus y así prevenir complicaciones graves de estas patologías, como la pancreatitis aguda. Caso clínico: Paciente de 30 años, con antecedentes familiares de DM2 y pancreatitis y personales de obesidad, que consulta el 08/06/12, derivada por medico dermatólogo luego de múltiples consultas a médicos clínicos, por xantomas eruptivos (confirmados por hallazgos histológicos) pruriginosos de tres meses de evolución, que progresan desde miembros inferiores hasta abarcar tronco y miembros superiores. Se solicita laboratorio y se constata suero lipémico con dosaje de triglicéridos 7.846mg/dl, glucemia 470 mg %, colesterol total 949mg/dl. Con diagnóstico inicial de Hipertrigliceridemia Familiar o Disbetalipoproteinemia tipo III asociada a DM, se inicia tratamiento con Gemfibrozil 900 mg/d y esquema de insulina NPH, aumentando posteriormente el gemfibrozil a 1200 mg/día. La paciente evoluciona con mejoría de las lesiones cutáneas y de los valores de laboratorio (a los 20 días de tratamiento: Triglicéridos 735 mg/dl, colesterol total 222, Glucemia 256 mg/dl). Con los resultados de los estudios solicitados a la paciente y a los familiares de primer grado se confirmara el diagnostico etiológico. Comentario: Resulta de particular interés el caso de una paciente que consulta por patología dermatológica sin otra signo sintomatología que haga sospechar asociación con dislipemia y en quien a posteriori por datos de laboratorio se diagnostica una Hipertrigliceridemia severa. FASEN 2012 CASOS CLÍNICOS GLUCAGONOMA METASTÁSICO, SEGUIMIENTO A LARGO PLAZO Ferrada P, Cuello L, Ojeda A, Savina M, Torres E, Herrera J, Bringa J. Servicio de Enfermedades Endocrino Metabólicas. Hospital Central de Mendoza, Mendoza, Argentina. 145 23 Introducción: El glucagonoma, tumor neuroendocrino funcionante de células alfa de los islotes pancreáticos, es una neoplasia infrecuente, con una incidencia de un caso cada 20 millones de habitantes. Objetivo: Presentar un caso de glucagonoma maligno metastásico, con un seguimiento de varios años, debido a lo infrecuente de esta entidad clínica. Caso: Paciente de 73 años de edad que comienza hace nueve años con astenia, pérdida de peso, diarrea y prurito en región perianal e inguinal. Inicialmente con glucemia normal. Se solicita tomografía axial computada de abdomen que muestra engrosamiento del cuerpo de páncreas. En otra institución se realiza punción aspirativa con aguja fina, con resultado negativo para citología neoplásica. En el seguimiento, aparición de tumor pancreático de crecimiento progresivo. Se indica cirugía, con negativa por parte del paciente. Tres años posteriores al hallazgo inicial de tumor pancreático, se diagnostica diabetes mellitus, con escasa respuesta a hipoglucemiantes orales y rápida necesidad de insulinoterapia (aprox. 1U/Kg insulina NPH), con gran inestabilidad en su control metabólico. Se agregan lesiones evanescentes eritematoescamosas con centro necrótico en dorso de pies y muslo, compatible con eritema necrolítico migratorio. Se realiza biopsia pancreática laparoscópica. Anatomía patológica: tumor neuroendócrino. Inmunohistoquímica: enolasa neurona específica, cromogranina A y glucagón positiva. Se realiza octreoscan con In111: intensa captación en tumor pancreático y múltiples nódulos hipercaptantes hepáticos. Diagnóstico: glucagonoma metastásico. Se inicia tratamiento con octeótrida (30 mg/mes) y oxido de zinc (50 mg/ día), con lo que el paciente recupera peso, mejora estado general, remiten lesiones cutáneas y cuadro diarreico. Durante los cuatro años iniciales de dicho tratamiento el paciente se mantiene en buen estado general con restitución a su actividad laboral, sólo apareciendo lesiones dermatológicas ante la demora en la dosis del agonista de somatostatina. En los últimos seis meses, a pesar del tratamiento indicado, reaparece la sintomatología inicial con exacerbación de las lesiones cutáneas aproximadamente a los veinte días de la aplicación del octeótride, por lo que se decide aumentar la frecuencia de la administración a 30 mg cada 15 días. Conclusión: En este caso la asociación entre tumor pancreático, diabetes y lesiones cutáneas específicas llevaron al diagnóstico de glucagonoma (elementos del denominado “síndrome glucagonoma”, hallado en el 70 % casos y descripto inicialmente por Becker en 1942), cuyo tratamiento con octeótrido y evaluación con octreoscan se basa en la presencia de receptores de somatostatina en tumores neuroendócrinos. Es frecuente la presencia de metástasis al momento del diagnóstico hasta en la mitad de los casos llegando este factor de malignidad a encontrarse en el 80 % de los pacientes. Existe acuerdo en que el tratamiento de elección del tumor primario es la resección quirúrgica; aunque como en este paciente, para las lesiones metastáticas la experiencia aún es reducida. El tratamiento diabetológico sugerido no difiere de otros tipos de diabetes, siendo la insulinoterapia la principal herramienta terapéutica. RAEM 2012. Vol 49 Nº Supl. 146 24 MANIFESTACIONES TIROIDEAS EN EL SÍNDROME HAMARTOMATOSO TUMORAL DEL PTEN Pastorino Casas V, Garuti R, Tosti R, Lutfi RJ, Faure E. Servicio de Endocrinología Hospital Churruca-Visca, CABA, Argentina. Introducción: El Síndrome de Cowden (SC) y el Síndrome de Bannayan-Riley-Ruvalcaba (SBRR) son expresiones fenotípicas distintas dentro de un mismo cuadro denominado Síndrome hamartomatoso tumoral del PTEN (SHTP) de herencia autosómica dominante, asociado a mutaciones en la línea germinal del gen de supresión tumoral PTEN. El diagnóstico es principalmente clínico ya que estas mutaciones se identifican en el 80 % de los SC y en el 60 % de los SBRR. El SC se caracteriza por macrocefalia, enfermedad de Lhermitte-Duclos (gangliocitoma displásico del cerebelo), lesiones viscerales y mucocutáneas hamartomatosas típicas, patología tiroidea y formación de neoplasias malignas, principalmente de mama y tiroides. El SBRR presenta macrocefalia, pólipos intestinales, lipomas, hemangiomas, máculas lentiginosas en pene y neoplasias. Caso clínico: Varón de 32 años en seguimiento por Gastroenterología por epigastralgia que persiste con omeprazol. Sin antecedentes personales de importancia. Antecedentes familiares: padre fallecido por cáncer de pulmón. VEDA y VCC: múltiples pólipos sésiles en esófago, estómago, duodeno, colon y recto. Anatomía patológica: ganglioneuromatosis intestinal. Es derivado a Endocrinología para estudio de patologías asociadas a ganglioneuromatosis. Refiere palpitaciones, disnea, temblor, disminución del apetito, constipación y pérdida de 17 kg de peso en 3 meses. Examen físico: clínicamente hipertiroideo; bocio multinodular; macrocefalia (perímetro cefálico de 63 cm, PC >97); facies dismórfica con hipertelorismo; testículos de 40-50 ml; pápulas blanco grisáceas en labios, hiperplasia gingival, lengua escrotal, triquilemomas faciales y en pabellón auricular, queratosis acral y lentiginosis en glande y prepucio (lesiones evaluadas por Dermatología). Laboratorio: TSH 0.01 μUI/ml (05-5),T4 total 19.4 μg/dl (4-12),T4 libre 2.78 ng/dl (0.8-1.8), T3 718 ng/dl (60200), ATPO 777.5 (+), ATG 1/400 (+), TRABS 60 % (h.15), LH 10.3 mUI/ml (2-12), FSH 6.6 mUI/ml (1-8), Testosterona total 15 ng/ml (2.7-17.3) y biodisponible 2.18 mmol/l (5-17), SHBG 180 nmol/l (7.2-100), PSA total 1.2 ng/ml (h.4) y libre 0.26 ng/ml (h.0.5), CEA 1 ng/ml (h.5), HCG 0 ng/ml, α feto proteína 2.3 ng/ml (h.10), Calcitonina 5 pg/ml (h.15), FAL total 405 UI/l (h.300) y ósea 55 UI/l (15-41), Deoxipiridinolina 19.5 nmol/mmol/ Cr (2.3-5.4), resto dentro de parámetros normales. Ecografía tiroidea: Múltiples nódulos heterogéneos, hiperecoicos con halo periférico y flujo tipo III, los de mayor tamaño en lóbulo derecho de 15 x 14 mm, en lóbulo izquierdo de 17 x 15 mm y en istmo de 10 x 8 mm, adenopatía yugular derecha de 6 mm de diámetro anteroposterior y ecoestructura alterada. Citología de nódulos: Categoría II de Sistema Bethesda (benigno). Citología y Tg PAAF de ganglio: negativas. Centellograma y captación tiroidea: Bocio multinodular con distribución heterogénea del radiotrazador e hipercaptación. TAC de cuello, tórax, abdomen y pelvis: Múltiples adenomegalias cervicales. Ecografía testicular: testículos aumentados de tamaño con múltiples imágenes difusas hiperecoicas menores a 3 mm compatibles con lipomatosis. Ecografía mamaria: normal. RMN de encéfalo: normal. Inicia Metimazol 30 mg/día y Propranolol 40 mg c/12 hs y se programa tiroidectomía total. Se solicita test genético en busca de mutación del PTEN (pendiente resultado). Durante evaluación familiar se detecta macrocefalia en hijo de 18 meses y en hija recién nacida. Conclusiones: El SHTP presenta una alta prevalencia de enfermedades tiroideas tales como bocio multinodular, tiroiditis autoinmune, hipotiroidismo, hipertiroidismo y cáncer papilar y folicular. Es importante el diagnóstico precoz del síndrome dado el alto riesgo de desarrollar tumores malignos. Se presenta este caso dada la rareza del mismo. FASEN 2012 Viernes 5 CASOS CLÍNICOS de octubre - Tiroides 1 - 25 y 147 26 - Salón Castellanos I - Coordinador: AM Orlandi FALLA HEPÁTICA E ICTERICIA EN PACIENTE CON HIPERTIROIDISMO Y HEMOCROMATOSIS Belingeri S*, Mana D*, Lopardi M*, Mezquida M**, Cazado E*. 25 Sección de Endocrinología Hospital de Agudos J.M. Penna*. Servicio de Gastroenterología Hospital de Agudos J. M. Penna** Pedro Chutro 3380, 1437 CABA, Argentina. daniela.mana@gmail.com Introducción: El hipertiroidismo es una patología frecuente de la práctica endocrinológica. La falla hepática es una complicación poco usual que se asocia al hipertiroidismo per sé o a su tratamiento con antitiroideos. Los mecanismos por los cuales se produce están aún en discusión. La hemocromatosis es una enfermedad autosómica recesiva caracterizada por el depósito multiorgánico de hierro, incluyendo la glándula tiroides. Caso clínico: Paciente de sexo masculino de 53 años que se internó por síndrome coledociano e insuficiencia hepática. Al interrogatorio refería pérdida de peso de 10 kilos y diarrea de 3 meses de evolución agregando en los últimos 15 días coloración amarillenta en piel y dolor abdominal. Como antecedentes presentaba enfermedad de Parkinson en tratamiento con agonistas dopaminérgicos los cuales había suspendido por su cuenta hacía 15 días. Negaba consumo de alcohol y drogas, sin otro antecedente patológico de importancia. Al examen físico adelgazado con ictericia en piel y mucosas. Petequias en ambos miembros inferiores y tronco. Abdomen doloroso en epigastrio e hipocondrio derecho con leve hepatomegalia. Temblor grueso y rigidez generalizada. Examen tiroideo normal y signos vitales conservados. Los resultados de laboratorio de rutina fueron Bilirrubina total 26.8mg % (h 1), Bilirrubina directa 21mg % (h 0.3), TGO 100 U/L (h 31), TGP 70 U/L (h 40), Gamma GT 70 U/L (h 49), Amilasa 11 U/L (h 100), TP 64 % (70-120), KPTT 40" (25-37), PCR 37 mg/L (0-5), VSG 89mm (h 20). El perfil tiroideo mostró TSH <0.01 uU/ml (0.27-4.2), T4 total >24.86 uU/dl (5.1-14.1) y T3 2.06 ng/ml (0.8-2.0). El perfil férrico arrojó ferremia y transferrina normales con niveles de ferritina de 1658 ng/dl (20-350). Serologías virales negativas así como brucelosis; descartándose también enfermedad de Wilson y patología tumoral como causa de colestasis. Ac Anti Receptor de TSH 8 % (>12) y ecografía tiroidea que mostró bocio heterogéneo difuso. Se inició tratamiento con metimazol 40mg/ día. La biopsia hepática presentó patrón compatible con cirrosis y hemocromatosis. El estudio genético fue negativo para los genes 63, 65 y 282 no descartando otras variedades menos frecuentes de dicha enfermedad. Al mes de iniciado el tratamiento con metimazol el paciente presentó mejoría del perfil tiroideo y del hepatograma con evolución favorable. Conclusión: Consideramos que en este paciente, con un deterioro hepático crónico e indolente por la hemocromatosis, el hipertiroidismo podría haber sido la causa de la falla hepática severa. En todo paciente que se presente con ictericia e insuficiencia hepática debería sospecharse hipertiroidismo como causa de dicho cuadro, aún en ausencia de síntomas y signos típicos de la enfermedad, debiendo confirmarse con la valoración bioquímica pertinente. RAEM 2012. Vol 49 Nº Supl. 148 26 CARCINOMA INSULAR DE TIROIDES EN UNA PACIENTE JOVEN Alarcón Lébérat H, Mega M, Benedettelli JC, Bonomi, A, Sosa Torres M, Carames, S, Falcón M, Perurena P. Hospital Interzonal de Agudos Evita Lanús, Servicio de Endocrinología y Medicina Nuclear endonuevita@intramed.net Tel. 011-4241-4051 al 59 int 313 Río de Janeiro 1910, Lanús 1824, Pcia de Buenos Aires, Argentina. El Carcinoma Insular de tiroides (CI) es un tumor pobremente diferenciado con baja frecuencia de presentación, descripto por primera vez en 1907 por Langhans; que en 1984 Carcangiu le da una nueva significación. El CI es mas frecuente en pacientes mayores de 50 años, con una relación mujer/hombre de 2 a 1. Presentamos un caso de una paciente de 37 años de edad, que presenta un difícil manejo quirúrgico. La paciente consulta en enero 2012 por una formación de reciente aparición, según refiere, por la cual es estudiada por su medico clínico constatándose dos formaciones nodulares de aproximadamente 27 mm cada una en lóbulo izquierdo, con función tiroidea normal. Al ser intervenida, la cirujana constata una formación fibrosa con gran adherencia a tráquea y esófago, por lo cual, al carecer de diagnostico realiza una resección parcial de la misma y decide esperar el informe anatomopatológico. El patólogo informa que por las características histologicas e inmunohistoquimicas conforma un cuadro compatible con un CI pobremente diferenciado. Luego de dicha cirugía se realiza TAC que informa agrandamiento heterogéneo de lóbulo izquierdo e itsmo y con desplazamiento de traquea y esófago; y escaso refuerzo post la administracion del contraste EV. Adenomegalias izquierdas supraclaviculares. No observándose lesiones pleuropulmonares ni mediastinales La ecografía consta formación en lobo izquierdo de 37 x 28 33 mm y adyacente a la misma otra de similares características de 18x19 mm y centellograma tiroideo TC99m donde se observa glándula tiroidea aumentada de tamaño lateralizada hacia la derecha por formación captante. Se realiza una segunda cirugía, cuyo informe patológico confirma la presencia del carcinoma insular y adyacente al mismo otra area de 15 x 10 mm correspondiente a un Carcinoma Papilar Variante Folicular. Post esta cirugía se repite el centellograma con tecnecio 99m observándose un area hipercaptante en proyección de lóbulo izquierdo e imagenes captantes en proyección de cadenas carotideas. Se realiza un ateneo debido a las dificultades técnicas quirúrgicas y de las imagenes donde se decidió aplicar una dosis ablativa de 150 mci de iodo 131. Se inicio a posteriori tratamiento con levotiroxina, y se realizaron imagenes post dosis donde se observo solo captación en similares areas ya descriptas. La ecografía muestra la resección total de lobulo derecho y un resto en proyección de lóbulo izquierdo, constatándose imágenes ganglionares, las cuales se proponen resecar quirúrgicamente si ello fuera posible Presentamos el caso por la edad de la paciente y sus características clínico-quirurgicas. FASEN 2012 Viernes 5 CASOS CLÍNICOS de octubre - Tiroides 2 y Misceláneas - 27 a 149 36 - Salón Castellanos II - Coordinadores: A Gauna CARCINOMA FOLICULAR DE TIROIDES DIAGNOSTICADO POR METÁSTASIS EN SISTEMA NERVIOSO CENTRAL Giacoia E, Donoso M, Corino M. 27 Servicio de Endocrinología y Metabolismo Hospital Nacional Prof. A. Posadas, CABA, Argentina. Introducción: En el carcinoma folicular de tiroides los sitios más frecuentes de diseminación son pulmón y hueso. La incidencia de MTS cerebrales es de 0.69 al 1.3%. Pueden tener invasión local y destrucción ósea subyacente. Su hallazgo empeora el pronóstico y la sobrevida de estos pacientes. Caso clínico: Mujer de 57 años con antecedentes de Diabetes tipo 2, y BMN seguida en otra institución, en tto con LT4 50mcg/d. Consultó a nuestro hospital por presentar tumoración palpable frontal derecha, de aprox. 2cm, duropétrea, de 3 meses de evolución, sin déficit neurológico. Se realizó TAC: “lesión ósea lítica en el hueso frontal sector derecho con componente intracraneal”, y RMN: “lesión extracerebral que compromete sector frontal derecho, área central hiperintensa en T1 que puede corresponder a hemorragia, con efecto de masa”. Evaluada por neurocirugía, se procede a resección del tumor con borde óseo infiltrado. Congelación: “tumor secundario de SNC con células redondas con diferenciación rosetoide”. Biopsia de cerebro: “MTS de CA tiroides que podría corresponder a folicular o papilar variante folicular, tejido óseo infiltrado por carcinoma de tiroides”. Es derivada a endocrinología en el postquirúrgico inmediato. Se confirma el diagnóstico de BMN, se palpan 2 nódulos duro-pétreos en lóbulo izquierdo de 2 y 3cm aprox. Clínicamente eutiroidea. LBT (bajo LT4 50/d): TSH 0.02 mU/l T4 9.4ug/dl T4L 1.03ng/dl TG 1387ng/ml ATG<2.2U/l ATPO 60. Presenta Rx tórax y ecografía abdominal sin particularidades. Laboratorio: Creat 0.5mg/dl FAL 295U/l Calcio 8.9mg/dl, resto normal. PAAF de tiroides: “Folículos y fragmentos de epitelio folicular con discreta alteración morfológica, oncológicamente dudoso”. Se efectúa tiroidectomía total, con resultado de biopsia: “LI: carcinoma folicular. LD: hiperplasia nodular”. Con TSH 54mU/L TG 25.8ng/ml ATG<2.2U/l se indica dosis I131 200mCi. RCT post dosis: “resto tiroideo”. Centellograma Oseo: “Area hipocaptante en zona postquirúrgica desplaquetada frontoparietal derecha”. Paciente poco adherente al seguimiento, sigue tomando LT4. Concurre 1 año después, exámen de cuello negativo. TAC cerebro control: “Hematoma epidural de forma lenticular en zona de craniectomía. Lesión ósea de aspecto benigno en tabla interna parietal izquierda, parénquima cerebral normal”. Recibe dosis de 100mci por resto en cuello, con TG (estimulada) 6.5ng/ml y ATG<2.2. A los 4 años de la cirugía la paciente comienza con cefalea, sin foco neurológico. Se solicitó RMN evidenciando 2 lesiones frontales. Se procede a resección de lesiones óseas adheridas a duramadre, informándose en la congelación “MTS carcinoma de tiroides en SNC”. Con TSH 64mU/l TG 3420ng/ml ATG<1U/l recibe dosis 200mci, en el RCT post dosis: “captación en calota”. Por persistencia de lesión cerebral con efecto de masa y deterioro clínico evidente se programa radioterapia externa (seguimiento conjunto con oncología). Recibe 5000 Gy. Inicia prednisona 40mg/d. Presenta TSH 0.14mU/l TG>464ng/ml ATG<0.1U/l. Se ausenta del seguimiento por 1 año. Regresa presentando tumoración en cuero cabelludo palpable fronto-parietal. Se asume persistencia de enfermedad y se indica 200mci. Ultimo RCT con lesiones óseas múltiples en columna dorsal, calota, cuello femoral izq, ²columna por dolor. Por su estado clínico no es pasible de quimioterapia. Se ausenta del seguimiento. Fallece en otro centro en mal estado general, 7 años después de la primera cirugía. Conclusión: La metástasis cerebral del carcinoma folicular de tiroides es infrecuente, la presentación única y la posibilidad de resección quirúrgica completa mejoran el pronóstico. En esta paciente el tiempo de sobrevida fue de 7 años luego del diagnóstico. RAEM 2012. Vol 49 Nº Supl. 150 28 LINFOMA PRIMARIO DE TIROIDES. PRESENTACIÓN DE UN CASO Ladenheim S, Filipponi A, Cononaco E. Unidad de Endocrinología. Hospital Pte. Perón. Anatole France 773 Avellaneda, Prov. Bs. As., Argentina Introducción: El linfoma primario de tiroides (LPT) es una patología poco frecuente, constituyendo menos del 2 % de todas las neoplasias tiroideas malignas y del 1 al 2,5 % de todos los linfomas. La mayoría de los mismos corresponden a linfoma no Hodgkin de células B, asociándose frecuentemente con tiroiditis de Hashimoto. Su diagnóstico genera dificultades, ya que la punción tiroidea pocas veces es de utilidad, debido a que la muestra obtenida para citología habitualmente es escasa e inespecífica con técnicas habituales. El tratamiento suele ser quirúrgico, en la mayoría de los casos, para lograr la confirmación diagnóstica, combinado posteriormente con ciclos de quimioterapia asociada o no a radioterapia local. Lo infrecuente que resulta este tipo de neoplasia linfoproliferativa en la glándula tiroides, de difícil diagnóstico clínico, nos motiva a la presentación de este caso. Caso clínico: Paciente de sexo femenino de 64 años de edad, oriunda de Paraguay, que consultó por primera vez en junio de 2009 por bocio de aproximadamente 3 años de evolución. En dicho momento, presentaba una glándula tiroides de 50 grs. multinodular sin poder delimitar borde inferior. Aportaba los siguientes estudios de laboratorio: TSH: 0,54uUI/ml, T4: 8,9ug/dl, T3: 116ng/dl, ATPO y ATG: negativos. Ecografía tiroidea: con lóbulo tiroideo derecho (LTD) de 50 x17 x 18 mm sin nódulo dominante y lóbulo tiroideo izquierdo (LTI) de 56 x 21 x 33 mm, heterogénea con zona multinodular. Centellograma que mostraba una glándula heterogénea, aumentada de tamaño, con mayor captación en LTI. Captaciones: 1° hora 5 % y 24 hs 12 %. TAC de cuello: tiroides aumentada de tamaño, multinodular, proyectándose al mediastino superior. Al año volvió a la consulta, con aumento de tamaño de la glándula tiroides asociado a disfonía y disfagia. Presentaba al exámen físico gándula tiroides de 100 gr. polinodular, con nódulo dominante en LTI de 4 cm de diámetro. Aportaba TSH: 0,53uUI/ml, T4: 10,27ug/dl, T3: 123ng/dl, rutina de laboratorio normal. Ecografia: que informó tiroides aumentada de tamaño, LTD con dos nódulos hipoecogénicos de 3 x 4 mm y de 10 x 6 mm. LTI: dos nódulos mixtos de 40 x 30 mm y de 20 x 17 mm. La punción tiroidea informó: extendidos hemáticos con presencia de cambios hiperplásicos en células foliculares. Debido al gran tamaño del bocio, síntomas compresivos y crecimiento progresivo, se indica tratamiento quirúrgico. Se realizó tiroidectomía total, siendo el informe anátomo-patológico: bocio multinodular presentando lesión focal de neoplasia maligna poco diferenciada parcialmente encapsulada e invasión focal de parénquima vecino (descartar neoplasia epitelial o linfoma). Por tal motivo, se efectuó estudio inmunohistoquímico correspondiendo a un linfoma no Hodgkin de células B periféricas o maduras subtipo linfoma de la zona marginal extraganglionar, linfoma MALT (tejido linfoide asociado a mucosas) de tiroides. Tras la confirmación diagnóstica, se derivó a la paciente al servicio de hematología, donde se le practicó el estudio de extensión en relación con dicho proceso linfoproliferativo; comenzando tratamiento quimioterápico con esquema CHOP-R (ciclofosfamida, adriamicina, vincristina, prednisona, rituximab). Conclusión: - El linfoma primario de tiroides, es una patología de baja frecuencia, siendo la punción tiroidea de poca utilidad para el diagnóstico, requiriendo en general en nuestro medio, obtener material histológico y realizar estudios inmunohistoquimicos para lograr su confirmación diagnóstica. - Destacamos que en este caso, el linfoma tiroideo subtipo MALT (que representa entre el 6 y el 27 % de los LPT), no está asociado a tiroiditis de Hashimoto. - Resaltamos la importancia de la sospecha clínica de malignidad, aun ante una citología negativa, para decidir la conducta terapéutica. FASEN 2012 CASOS CLÍNICOS RESOLUCIÓN QUIRÚRGICA POR VÍA CERVICAL DE BOCIO NODULAR CON GRAN PROLONGACIÓN ENDOTORÁCICA Calabrese MC*, Abalovich M*, Alcaraz G*, Vázquez A*, Frydman M*, Desiderio A, Gutiérrez S* 151 29 *Grupo de Trabajo “Tiroides”, División Endocrinología y Departamento de Cirugía, Hospital Carlos G. Durand. CABA, Argentina. Mujer de 73 años que consulta en abril del 2002 por bocio nodular de 10 años de evolución. A la palpación: nódulo de 6 x 5 cm que abarca istmo y se prolonga a ambos lóbulos a predominio derecho. No se palpa borde inferior. Laboratorio basal (8/2002): TSH <0.01 mUI/L; T4 11.3 μg/dL; aTPO y aTg Neg; TRAb 15%. TAC de cuello y tórax (8/2002): Gran aumento de la glándula tiroides que se insinúa por el opérculo torácico y comprime la tráquea. Bajo tratamiento con MMI 5 mg/d previo a cirugía: TSH <0.01; T4 7.1; T3 161 ng/dL. Se suspende cirugía por crisis HTA. Regresa a la consulta en 12/2007 sin MMI desde el 2003. TAC de cuello y tórax (12/2006): tiroides aumentada de tamaño, deformada, heterogénea, parcialmente calcificada multinodular. Extensa formación cervicotorácica que ejerce efecto compresivo sobre tráquea y esófago desviándolos hacia la izquierda y adelante. Laboratorio: TSH 0.09; T4 13.3; T3 204; aTPO neg. PAAF: bocio coloide. Se inicia MMI 5 mg/d. Rechaza cirugía. Vuelve a la consulta en 08/2009 por presentar ortopnea y estridor laríngeo al flexionar la cabeza. Continuó tomando MMI 5 mg/d. Tiroides con progresión del bocio y una formación policíclica retroECM izquierda que impresiona palpatoriamente compatible con cadena adenopática, no evidenciable por ecografía. PAAF: Formación policíclica izquierda: tejido tiroideo Tg-PAAF >30000 ng/mL. Nódulo ístmico: bocio coloide. Bajo MMI 5mg: TSH 0.36; T4 7; T3 132; Calcitonina <2 pg/mL; PTH 28 pg/mL; Cai 5.39 mg/dL. EFR: alteración ventilatoria restrictiva y obstructiva leve. RNM de cuello y mediastino: Lesión de contornos irregulares de 13x7x8.5 cm, en íntimo contacto con los ECM y pedículos yugulocarotídeos con proyección mediastinal hasta plano prevertebral y grandes vasos. Deformación y desplazamiento anterolateral izq. de la tráquea en contacto con esófago. Tiroidectomía total por abordaje cervical con maniobras digitales para extraer tejido del plano mediastinal prevertebral. (26/10/2009). Biopsia por congelación: bocio coloide. No se visualizan ADP. Prueba hidráulica que descarta fuga de aire. Se extuba exitosamente. No presentó traqueomalacia ni hipoparatiroidismo. AP: Bocio multinodular hiperplásico. Carcinoma papilar de 1 cm sin invasión de la cápsula tiroidea. Laboratorio Preablación: TSH: 34.5; T4 1.76; Tg 9.36; AcTg: < 20. Captación con 500 μCi I131: 3 - 5 %. Centellograma positivo en línea media sobre la cicatriz quirúrgica. Se administran 100 mCi I131. BCT post dosis ablativa: positivo en línea media. Bajo LT4, subjetivamente bien, sin disnea. Ecografía de cuello: no se observa remanente glandular en la lodge quirúrgica. Granuloma calcificado de 3 mm en territorio paratraqueal derecho, sin ADP. Motiva esta presentación la exitosa resolución quirúrgica por vía cervical de un bocio polinodular, en paciente añosa con severo compromiso respiratorio. Se optó por la radioablación ante los antecedentes reiterados de mala compliance. Prequirúrgico Postquirúrgico RAEM 2012. Vol 49 Nº Supl. 152 30 ESTUDIO FAMILIAR DE PACIENTES CON DEFICIENCIA DE GLOBULINA FIJADORA DE TIROXINA Parma, R, Peralta, E, Chiarpenello, J, Pena, C. Hospital Provincial del Centenario, Urquiza 816 PB, 2000 Rosario, Prov. Santa Fe, Argentina. Tel.: 0341-4474791 penacarolinalujan@hotmail.com, parmaricardo7@gmail.com Objetivo: Estudio de paciente índice y su familia con deficiencia de globulina fijadora de tiroxina. Material y métodos: Se estudio paciente índice de sexo masculino, 28 años de edad, empleado, motivo de consulta: cansancio, fatiga, astenia, cefaleas, de 10 meses de evolución; antecedentes familiares, madre hipotiroidea en tratamiento con levotiroxina de larga data. Se solicitan exámenes complementarios al paciente índice: laboratorio hormonal: TSH: 1.96, T4T: 2.3 (5.3-11.1), T3T: 62 (80-220), T4L: 1.57 (0.93-1.7), ATA Y ATPO: NEGATIVOS. Por lo expuesto se plantea diagnostico diferencial: HIPOTIRODISIMO SECUNDARIO Y DEFICIENCIA DE GLOBULINA FIJADORA DE TIROXINA. Para confirmar se solicita, megatest hipofisario, resonancia de hipófisis. Ejes hipofisarios normales, resonancia de hipófisis: normal. Por lo que se decide dosaje de TBG. El dosaje de globulina fijadora de tiroxina confirma la deficiencia, por lo que se decide estudio familiar, por la herencia confirmada ligada al cromosoma x. Resultados: El dosaje de globulina fijadora de tiroxina del paciente índice y de la familia, de 2 miembros( madre y paciente índice), confirma la deficiencia de globulina fijadora de tiroxina. Es de destacar que, como hallazgo, el padre del paciente presenta una Tiroiditis de Hashimoto confirmado por laboratorio hormonal, anticuerpos y ecografía de tiroides, es decir, un hipotiroidismo primario. La TBG (thyroxine-binding-globulin) es un glicoproteína acida que consta de 1 sola cadena polipeptídica de 395 AA y 4 cadenas de oligosacáridos.Es sintetizada por el hígado. La capacidad de fijación de T3 total circulante (70 %) y T4 total circulante (75 %), es equivalente a su concentración. La concentración serica de TBG en sujetos normales es: 1.5mg/dl ( 0.27Umol/l). La TBG esta codificada por una sola copia del gen localizado en el cromosoma X(locus: Xq 22.2) Los déficit de TBG son frecuentes. Las mutaciones en el gen de TBG lleva al desarrollo de diversas variantes del la proteína globulina fijadora de tiroxina que por su concentración puede crear 3 fenotipos de las anomalías de TBG: DEFICIENCIA COMPLETA, PARCIAL O EXCESO DE TBG. Herencia del cromosoma X relacionada con varones homocigotos afectados y mujeres portadoras heterocigotos con valores intermedios para T4 TOTAL Y TBG. Se caracterizan clínicamente por EUTIROIDISMO, con T4 T y T3T BAJAS CON T4 L y T3L y TSH: NORMALES. Es causada por una terminación prematura de la traducción C-terminal de la alteración de AA o una sustitución de AA, resultado: FRACASO DE LA SECRECION. Se adjunta en la siguiente tabla los datos familiares: Bibliografía: - Katarzyna Lacka, Teresa Nizankowska, Agnieszka Ogrodowick, and Jan K. Lacki,: A Novel Mutatio(del 1711G) in the TBG Gene as a, Cause of Complete TBG Deficiency, Thyroid, 2007 Nov: 17(11): 1143-1146. - Schussler GC 2000 The thyroxine-binding proteins. Thyroid 10:141-149. - Nicoloff JT, Dowling JT, Patton DD, Inheritance of decreased thyroxine-binding by the thyroxine-binding globulin. J Clin Endocrino Metab 24:294-298. FASEN 2012 CASOS CLÍNICOS Patologías tiroideas asociadas: discrepancias clínico-bioquímicas Gattelli AC, Gallinger C, Prener P. Hospital Interzonal Especializado en Agudos y Crónicos “San Juan de Dios” de La Plata. Bs. As. Argentina. 153 31 Introducción: Los trastornos mixtos en la función tiroidea resultan un desafío diagnóstico. La enfermedad autoinmune tiroidea es una causa frecuente de disfunción de la glándula y en raros casos se asocia a procesos que alteran la homeostasis tiroidea, hereditarios o adquiridos, como son la resistencia a hormonas tiroideas, modificaciones cuali o cuantitativas en las proteínas de transporte o presencia de autoanticuerpos anti T3 o T4. El diagnóstico, en estas circunstancias, requiere de un análisis cuidadoso de los datos clínicos y de laboratorio. Objetivos: presentar un caso de hipotiroidismo de etiología, parámetros bioquímicos y clínica controversial. Caso clínico: paciente femenino de 36 años de edad, proveniente de la ciudad de La Plata, Pcia. de Bs As, que consulta al servicio de Dermatología del hospital por caída abrupta y marcada de cabello. El profesional médico constata la alopecia y durante el interrogatorio la paciente refiere sequedad de piel, aumento de peso, alteración del sueño, nerviosismo, irritabilidad emocional, cansancio, constipación y alteración de los ciclos menstruales. Antecedentes familiares: madre hipotiroidea, dos primos hermanos maternos tiroidectomizados, una prima hermana materna operada por carcinoma medular de tiroides y hermanos que no refieren sintomatología ni estudios hormonales a la fecha. Se le solicita dosaje de hormonas tiroideas, TSH y anticuerpos antitiroideos. El resultado (5/1/10) arroja valores patológicos pero discordantes con la clínica por lo cuál el laboratorio extrae una nueva muestra (18/01/10) para confirmar. Los resultados de laboratorio obtenidos durante todo el seguimiento se muestran ordenados cronológicamente en la Tabla 1. Estudios de imágenes: TAC de silla turca normal, ecografía de tiroides con hallazgos compatibles con tiroiditis. En el mes de marzo comienza tratamiento con 50mg de levotiroxina/día que luego aumentó a 75mg/día. Un año más tarde (1/06/2011) el endocrinólogo solicita un test de estímulo con TRH obteniéndose TSHbasal: 10,24, TSH30min: 136, ΔTSH: 126, PRLbasal: 15,2, PRL30min: 62,4. A la fecha el esquema terapéutico consiste en 4 tomas a la semana de 75mg Levotiroxina/día y 3 tomas de 100mg/ día. La paciente se encuentra clínicamente eutiroidea. Tabla 1. Resultados del período de estudio. Las unidades de expresión son: T3 ng/dl, T4 µg/dl, T4L ng/dl, TSH µUI/ml, anti-Tiroglobulina (a-Tg) UI/ml, anti- peroxidasa (a-TPO) UI/ml, Prolactina (PRL) ng/ml. 5/1/10 18/1/10 2/104/106/105/1/11 17/1/11 6/11 9/115/12 VR T3 n(nssng/dl ng/dl T4 T4libre TSH μUi/ml a-Tg a-TPO 135 159 132 155 125 157 130 131 70-170 11.312,5 15,8 12,813,5 15,0 14,4 4,5-12,5 2,1 2,4 1,7 1,92,12 2,38 2,18 1,85 1,79 1,81 0,8-1,9 1,08 8,55 6,44,74,7 4,276,64 10,25,06,50,4-4,0 ND ND ND 0-40 608 797894 0-35 Discusión: basándonos en el historial bioquímico y la clínica postulamos la coexistencia en esta paciente de dos mecanismos fisiopatológicos. La presencia de valores elevados de anti-TPO al inicio del cuadro con ecografía complementaria se asocian a enfermedad tiroidea autoinmune. Además, la hiperrespuesta de la prueba de TRH es compatible con hipotiroidismo primario. Lo controversial se observa en los niveles persistentes de T4 y T4 libre (T4L) elevados o en el límite superior de normalidad, TSH no suprimida y respuesta alterada a TRH aún cuando la paciente se encuentra con terapia sustitutiva hormonal. Estos últimos hallazgos sugieren la presencia de un inhibidor/bloqueador, una modificación en la afinidad de la albúmina por la T4 o algún tipo de resistencia sistémica a la acción de las hormonas tiroideas. Considerando los antecedentes familiares las hipótesis son: resistencia hereditaria que la mantuvo eutiroidea hasta que desarrolló una tiroiditis; un cuadro de tiroiditis autoinmune con expresión de autoanticuerpos contra T4 y T3 que interfieren in vivo e in vitro en las técnicas de medida; Hipertiroxinemia Disalbuminémica Familiar sumada a una tiroiditis autoinmune. Conclusión: para un abordaje más certero proponemos la realización de estudios de biología molecular que evalúen mutaciones en el receptor ß de hormonas tiroideas y en el gen de albúmina, como la búsqueda de anticuerpos anti-T4 y T3 circulantes. Bibliografía - Bernal J. Síndromes de resistencia a hormonas tiroideas. Endocrinol Nutr.2011;58(4):185-196. - Sabet A., Pallotta J. Dichotomous responses to thyroid hormone treatment in a patient with primary hypothyroidism and thyroid hormone resistance Thyroid.2011 May; 21(5):559-61. RAEM 2012. Vol 49 Nº Supl. 154 32 EFECTO ADVERSO GRAVE DEL TRATAMIENTO MÉDICO DEL HIPERTIROIDISMO: AGRANULOCITOSIS Cuello L, Ferrada P, Ojeda A, Savina M, Torres E, Bringa J. Servicio de Enfermedades Endocrinometabólicas. Hospital Central de Mendoza. Mendoza, Argentina. Introducción: El cáncer de paratiroides es poco frecuente, se encuentra en el 1 % de los hiperparatiroidismos primarios y suele presentarse con hipercalcemia severa. La cirugía es el tratamiento de elección, y la resección completa del tumor en la cirugía inicial es crucial en la evolución posterior de los pacientes. Material y métodos: presentamos 2 pacientes con motivos de consulta diferentes con carcinoma de paratiroides Caso 1: paciente de sexo femenino de 72 años de edad que en TC de cuello de control post neumonía, se detecta nod de 28.3mm en lob. Derecho tiroideo con calcificaciones en su interior, con áreas solidas y quísticas se extiende hacia abajo, desplazando la tráquea y el esófago a la izq. No adenopatías. Laboratorio: Cas 10.2, creat 1.01, TSH 3.71, atpo 99.7, atg neg Ecog tiroidea LDER aum de tamaño nod de 10 x 9 e imagen compleja de 50 x 27 con prolongación endotoracica, LIZQ 30 x 14 x 12 Paaf: sospechosa de proliferación folicular, Bethesda IV. Cirugía: lobectomía derecha mas istmectomia nod tiroideos de 1 y 0.5 cm, adherido al lóbulo derecho nódulo de 2.5 cm. Anat. Patológica: tiroides bocio nodular, nódulo: carcinoma paratiroideo con invasión de tejidos blandos peri glandulares e invasión de vasos linfáticos. Caso 2: paciente de sexo femenino de 67 años de edad que consulta por hipercalcemia detectada en un laboratorio de rutina. Tenía antecedentes de Hipotiroidismo y bocio multinodular. Laboratorio: Cas 11.5, Cai 7, creat 1, Cau 24 hs 249, Ca/creat 24 hs 0.41, PTH 199, FAL 308 Ecografía tiroidea: LDer nod de 36 x 12 x 13 en 1/3 sup, LIzq nod de 19 x 9 x 17 en vértice y nódulos de 13 x 11 en base Centellograma de paratiroides: área hipercaptante en polo inferior del lob derecho con alta probabilidad de corresponder a adenoma paratiroideo o nódulo tiroideo autónomo. DMO de raquis L1-4 1.393g/cm T score 1.8.¡ CFD: 0.876g/cm T score -1.6 Ecografía renal: sin litiasis Paaf tiroidea: de los 3 nódulos tiroideos compatibles con hiperplasia nodular. Cirugía: Para tiroidectomía superior derecha y Tiroidectomía total. Anatomía Patol: Paratiroidectomía superior derecha: Carcinoma paratiroideo que compromete la capsula e infiltra focalmente los tejidos peri glandulares, no se observa invasión linfática, ni vascular. Biopsia de paratiroides superior izquierda: normal Tiroidectomía total: Bocio coloide multinodular. Discusión: La media de edad de presentación del cáncer paratiroideo es en la 5ª década (mas jóvenes que en hiperparatiroidismo primario por patología benigna) en algunos casos se asocia a adenoma paratiroideo y a hiperparatiroidismo secundario a enfermedad celiaca en estadios terminales de la insuficiencia renal; también asociado a radioterapia de cuello. Los niveles de calcemia suelen estar muy elevados con Cas mayores de 15 mg/dl y los niveles de PTH mayores de 1000pg/ml. Nuestras dos pacientes no se presentaron como es referido en la literatura, el caso 1 se presento con normocalcemia, pero con signos de invasión local y la segunda presento moderada hipercalcemia sin osteoporosis ni litiasis renal. La sospecha clínica prequirurgica es fundamental ya que la técnica quirúrgica es diferente. En estas dos pacientes se realizo la técnica quirúrgica adecuada por la coexistencia de patología nodular tiroidea en una, y por la sospecha clínica de malignidad tiroidea por la invasión local del tumor en la otra. Si bien la prevalencia de carcinoma de paratiroides es poco frecuente debe tenerse presente en pacientes que van a cirugía por hipercalcemia moderada o ante una masa tumoral en el cuello. FASEN 2012 CASOS CLÍNICOS DIFERENTE PRESENTACIÓN CLÍNICA DE DOS PACIENTES CON CÁNCER DE PARATIROIDES Premrou MV, Paizal AM, Maffei L. 155 33 Consultorios Asociados de Endocrinología. Cerviño 3375 1er piso oficina 2. 1425 CABA, Argentina. valepremrou@hotmail.com Introducción y objetivos: El cáncer de paratiroides es poco frecuente, se encuentra en el 1 % de los hiperparatiroidismos primarios y suele presentarse con hipercalcemia severa. La cirugía es el tratamiento de elección, y la resección completa del tumor en la cirugía inicial es crucial en la evolución posterior de los pacientes. Material y métodos: presentamos 2 pacientes con motivos de consulta diferentes con carcinoma de paratiroides. Caso 1: paciente de sexo femenino de 72 años de edad que en TC de cuello de control post neumonía, se detecta nod de 28.3mm en lob. Derecho tiroideo con calcificaciones en su interior, con áreas solidas y quísticas se extiende hacia abajo, desplazando la tráquea y el esófago a la izq. No adenopatías. Laboratorio: Cas 10.2, creat 1.01, TSH 3.71, atpo 99.7, atg neg Ecog tiroidea LDER aum de tamaño nod de 10x9 e imagen compleja de 50 x 27 con prolongación endotoracica, LIZQ 30 x 14 x 12 Paaf: sospechosa de proliferación folicular, Bethesda IV Cirugía: lobectomía derecha mas istmectomia nod tiroideos de 1 y 0.5 cm, adherido al lóbulo derecho nódulo de 2.5 cm Anat. Patológica: tiroides bocio nodular, nódulo: carcinoma paratiroideo con invasión de tejidos blandos peri glandulares e invasión de vasos linfáticos. Caso 2: paciente de sexo femenino de 67 años de edad que consulta por hipercalcemia detectada en un laboratorio de rutina. Tenía antecedentes de Hipotiroidismo y bocio multinodular. Laboratorio: Cas 11.5, Cai 7, creat 1, Cau 24 hs 249, Ca/creat 24 hs 0.41, PTH 199, FAL 308 Ecografía tiroidea: LDer nod de 36 x 12 x 13 en 1/3 sup, LIzq nod de 19 x 9 x 17 en vértice y nódulos de 13 x 11 en base. Centellograma de paratiroides: área hipercaptante en polo inferior del lob derecho con alta probabilidad de corresponder a adenoma paratiroideo o nódulo tiroideo autónomo. DMO de raquis L1-4 1.393g/cm T score 1.8 CFD: 0.876g/cm T score -1.6 Ecografía renal: sin litiasis Paaf tiroidea: de los 3 nódulos tiroideos compatibles con hiperplasia nodular. Cirugía: Para tiroidectomía superior derecha y Tiroidectomía total. Anatomía Patol: Paratiroidectomía superior derecha: Carcinoma paratiroideo que compromete la capsula e infiltra focalmente los tejidos peri glandulares, no se observa invasión linfática, ni vascular. Biopsia de paratiroides superior izquierda: normal Tiroidectomía total: Bocio coloide multinodular. Discusión: La media de edad de presentación del cáncer paratiroideo es en la 5ª década (mas jóvenes que en hiperparatiroidismo primario por patología benigna) en algunos casos se asocia a adenoma paratiroideo y a hiperparatiroidismo secundario a enfermedad celiaca en estadios terminales de la insuficiencia renal; también asociado a radioterapia de cuello. Los niveles de calcemia suelen estar muy elevados con Cas mayores de 15 mg/dl y los niveles de PTH mayores de 1000pg/ml. Nuestras dos pacientes no se presentaron como es referido en la literatura, el caso 1 se presento con normocalcemia, pero con signos de invasión local y la segunda presento moderada hipercalcemia sin osteoporosis ni litiasis renal. La sospecha clínica pre quirúrgica es fundamental ya que la técnica quirúrgica es diferente. En estas dos pacientes se realizo la técnica quirúrgica adecuada por la coexistencia de patología nodular tiroidea en una, y por la sospecha clínica de malignidad tiroidea por la invasión local del tumor en la otra. Si bien la prevalencia de carcinoma de paratiroides es poco frecuente debe tenerse presente en pacientes que van a cirugía por hipercalcemia moderada o ante una masa tumoral en el cuello. RAEM 2012. Vol 49 Nº Supl. 156 34 TRATAMIENTO CON LIRAGLUTIDE EN PACIENTE CON PANHIPOPITUITARISMO Y DIABETES MELLITUS Ferrada P., Cuello L., Ojeda A., Savina M., Torres E., Herrera J., Bringa J. Servicio de Enfermedades Endocrinometabólicas. Hospital Central de Mendoza. Argentina. Introducción: la presencia de diabetes en un paciente con déficit hormonal hipofisario representa un verdadero desafío al buscar un adecuado control metabólico. El déficit de somatotrofina determina una mayor frecuencia de obesidad visceral, insulinorresistencia y dislipemia, con aumento de la mortalidad cardiovascular en estos pacientes. El síndrome metabólico ante déficit de GH se encuentra en hasta la mitad de los casos. La perdida de hormonas contrarreguladoras de insulina (cortisol, hormona de crecimiento), confiere a estos pacientes un mayor riesgo de hipoglucemias ante la coexistencia de diabetes y la instauración de un plan de insulinoterapia intensificada. Objetivo: presentar un caso de un paciente diabético panhipopituitario, con dificultad para manejo del control glucémico y mejoría del mismo ante la indicación de liraglutide como tratamiento complementario para su diabetes. Caso clínico: varón de 46 años de edad. Antecedentes: hipopituitarismo (macroprolactinoma tratado con cirugía y radioterapia a los 20 años de edad), diabetes mellitus tipo 1 lábil sin reserva insulínica (Ac antiGAD 0.94 U/ml, VR hasta 1.00; Péptido C < 0.10 ng/ml, VR 1.1-5.00; ICA-Ac anticélula β pancreática 40 JDF, VR < 10) de 8 años de evolución. Ante la falta de controles médicos y tratamiento adecuado de su función hipofisaria hasta los 40 años de edad, el paciente desarrolló síndrome metabólico, el cual mejoró considerablemente al iniciar reemplazo androgénico y del déficit de hormona de crecimiento. A partir de la dificultad para conseguir un adecuado control glucémico se fueron realizando múltiples modificaciones de la insulinoterapia, sin lograr mejorar la importante variabilidad glucémica que presentaba, con un deterioro en la calidad de vida justificado por frecuentes hipoglucemias sintomáticas que han requerido internaciones en numerosas ocasiones. Esto motivo el inicio de tratamiento con liraglutide 5 meses previos, con mejoría del perfil metabólico, disminución de la variabilidad glucémica, menor requerimiento de dosis de corrección preprandial con insulina aspártica ante la utilización de 0.6 mg cada 12 horas del agonista de GLP-1. Tratamiento hormonal sustitutivo actual: levotiroxina 150 mg/día, hidrocortisona 20 mg/día, enantato de testosterona 100 mg c/15 días, hormona de crecimiento 0.4mg/ día. Insulinoterapia: insulina glargina, insulina aspártica (según monitoreo y conteo de hidratos de carbono), liraglutide 0.6mg/12hs. Laboratorio actual: HbA1c 7.2 % (valores previos > de 9 %). Discusión: la utilización de liraglutide como tratamiento de la diabetes tipo 1, es un hecho no frecuente del cual figuran limitadas experiencias en la literatura mundial actual. Con este tratamiento en un grupo de pacientes diabéticos tipo 1 con y sin reserva insulínica, se observó una reducción de la dosis de insulina, sin empeoramiento de control glucémico incluso con menor variabilidad y reducción de peso en todos los pacientes bajo el agonista GLP-1. También se ha publicado un reporte de 3 casos de pacientes con diabetes 1 y MODY con similares resultados. En el grupo de diabéticos tipo 2, donde este tratamiento tiene más experiencia, además de lo anterior se cita la mejoría en el perfil lipídico, tensión arterial y la calidad de vida de los sujetos tratados, todos factores de interés a tener en cuenta en nuestro paciente debido a su patología de base con tendencia a desarrollo de síndrome metabólico. Probablemente el mecanismo implicado en pacientes con baja reserva insulina incluya a la regulación del glucagón como principal actor en esta mejoría observada. FASEN 2012 CASOS CLÍNICOS DESAPARICIÓN DE MACROSOMATOTROPINOMA EN UNA PACIENTE CON ACROMEGALIA LUEGO DE TRATAMIENTO CON ANÁLOGOS DE SOMATOSTATINA Bamberger E. PAMI Delegación - Patricias Mendocinas 812 157 35 Ciudad de Mendoza. Mendoza. Argentina. Los análogos de la somatostatina son eficaces para lograr la mejoría o remisión bioquímica de la acromegalia y también para obtener la reducción del tamaño tumoral, aunque la desaparición completa del adenoma ha sido comunicada sólo en pocos casos. Una mujer de 38 años, consultó en 2006 por síntomas de acromegalia: artralgias, edemas, dolores óseos y aumento de partes acrales desde los 27 años. Presentaba además, prognatismo, aumento del surco nasogeniano, oligomenorrea, galactorrea serolactescente, manos suculentas, tiroides irregular, macroglosia, hirsutismo en región sacra; calzado nº 44 (previo 39), peso actual 95 kg (habitual 75) con talla 165 cm y se hallaba normotensa. Una resonancia magnética nuclear (RMN) demostró un macroadenoma hipofisario de 22 mm de diámetro máximo con extensión supraselar, en contacto con el quiasma óptico e invasión del seno esfenoidal. La medición de GH fue de 47.7 ng/ml (vn <5.0) y la de PRL 73.5 ng/ml (vn < 24). Se indicó cabergolina inicialmente y fue derivada a Neurocirugía donde dicho tratamiento fue continuado sin controles bioquímicos, abandonando el seguimiento endocrinológico. Cuatro años más tarde, la paciente fue nuevamente enviada a Endocrinología por presentar crecimiento del tumor pituitario, ahora de 22 x 29 mm en la RMN, con invasión de senos cavernosos y compresión quiasmática. El estudio campimétrico reveló hemianopsia bilateral superior con importante disminución visual en OI y visión borrosa en OD. Una ecografía abdominal demostró la presencia de colelitiasis y una ecografía tiroidea evidenció un bocio polinodular; la PAAF de nódulos dominantes fue compatible con bocio coloide. La medición de GH fue de 7.7 ng/ml y la de IGF-1: 725 ng/ml (vn <267) Nuevamente la paciente discontinuó sus controles durante un año y, luego de su retorno a Endocrinología en 2011, la RMN mostró un macroadenoma de 33 x 26 x 32 mm. El examen oftalmológico mostró signos de atrofia del nervio óptico. Su GH fue 18.3 ng/ml e IGF-1: 615 ng/ml (vn <264). Durante este lapso desarrolló HTA y diabetes tipo 2. La paciente fue finalmente operada por vía transciliar en mayo de 2011 pero la RMN post-quirúrgica no evidenció cambios respecto de la pre-quirúrgica. El estudio anatomopatológico fue informado como: “fragmentos tisulares disgregados constituidos por sustancia blanca; no se reconoce neoplasia en los cortes estudiados”; la IGF-1 obtenida 3 meses más tarde fue de 650 ng/ml (<284). Neurocirugía indicó radiocirugía pero Endocrinología la contraindicó debido al tamaño del tumor y a su cercanía con el quiasma. Se inició tratamiento con análogos de la somatostatina recibiendo una inyección de octreotida LAR 30 mg, seguida de 5 aplicaciones mensuales de lanreotida 120 mg. La paciente notó mejoría de las artralgias y de su visión y se constató normoglucemia. Una RMN obtenida luego de 6 aplicaciones de análogos evidenció aumento de la silla turca ósea e imágenes que probablemente correspondieran a restos glandulares y lesión residual en el piso de la misma, con realce heterogéneo luego de la administración del contraste, no observándose la lesión expansiva previa, e invaginación del parénquima frontal a través de la cisterna supraselar, ocupando la silla turca. Los controles bioquímicos no evidenciaron, por el contrario, normalización de IGF-1 (545 ng/ml, a 6 meses de iniciado el tratamiento). Esta presentación ilustra sobre las dificultades en el manejo de una paciente acromegálica, derivadas de diversas razones que frecuentemente impiden, en la vida real, atenerse a los algoritmos sugeridos en diversos consensos internacionales. En este caso, el empleo de análogos de la somatostatina, principalmente lanreotida, condujo a la desaparición de la masa tumoral en seis meses, aunque con persistencia de actividad bioquímica. RAEM 2012. Vol 49 Nº Supl. 158 36 DIAGNÓSTICO DE ENFERMEDAD DE PAGET EN VARONES HIPOGONÁDICOS Calletti FS, Lalosa S, Salerni H, Karlsbrum S, Aszpis S, Levalle O. Div. Endocrinología, Htal. Durand., Díaz Vélez 5044, (405) CABA, Argentina. endodurand@fibertel.com.ar Tel. 4982-5212 La enfermedad de Paget (EP) es un desorden del remodelado óseo poco frecuente: tiene una prevalencia del 4% a partir de los 50 años. Se presentan dos casos de asociación de hipogonadismo secundario (HS) y EP. CASO 1: Varón de 24 años que consultó por falta de desarrollo puberal, antecedentes de hidrocefalia, criptorquidia bilateral, astenia y libido disminuida. Examen físico: hábito eunucoide, vello pubiano ginecoide, testículos no palpables. Resultados de estudios patológicos: FSH 0.27mUI/ml (1.5-7), LH 0.2 mUI/ml (1.1-12), Testosterona(T) 0.15 ng/ml (3-9), Estradiol (E) <15 pg/ml; 25(OH)D 13.7 ng/dl (> 40), FAL 432 UI/l (60-270); edad ósea 18 años; DMO: Osteoporosis L2-L4: Z-score -4.7. Se inició terapia de reemplazo hormonal (TRH) con enantato de testosterona, ergocalciferol y citrato de calcio, con mejoría de su HS. A los 6 meses se constató T 4.4 ng/ml, FAL 1.000 UI/l, FAO 115 UI/l (4.3-20) y centellograma óseo con fijación en hombros, rodillas y tarsos y distribución irregular en calota. Evolución: Se rotó el reemplazo androgénico a T gel y a los 4 meses de eugonadismo, evidenció FAL 690 UI/l y FAO 71 UI/l. Por la criptorquidia, se inició tratamiento con hCG, observándose a los 6 meses testículos en bolsas por ecografía. A los 10 meses se verificó FAL 446 UI/L, FAO 55.3 UI/l. Reinició T gel. A los 5 años: FAL 439 UI/l, FAO 35 UI/l; RX cráneo: áreas algodonosas y deflecadas e hiperostosis frontal. A los 6 años: FAL 427 UI/l, FAO 38.2 UI/l (Centellograma óseo: hiperfijación difusa en calota a predominio frontal; Rx cráneo: engrosamiento de la tabla y levemente algodonoso, Rx de pelvis: cierre de cartílago de cresta ilíaca, artrosis coxofemoral. A 7 años: PTH 39.4 pg/ml (15-65), 25(OH)D 38 ng/dl, FAL 410 UI/l, FAO 33.6 UI/l; A lo largo del seguimiento la DMO aumentó 30 %; L2-4 Z -2.3. Estabilizadas FAL/FAO con cierre total del cartílago de crecimiento se inicia tratamiento para EP con risedronato 35mg bisemanal, presentando a los seis meses una FAO de 29.8 UI/l. CASO 2: Varón de 42 años que consultó por esterilidad de 10 años de evolución, con antecedentes de hipogonadismo, azoospermia, cirugía por criptorquidia bilateral, infantilismo sexual, fiebre urliana, e hiposmia. Examen físico: escaso vello corporal y testículos no palpables. Resultados de estudios patológicos: espermograma (volumen 0,5 ml; pH 6,5; fructosa no dosable, azoospermia pos centrifugación); FSH 2.79 mUI/ml, LH 0.7 mUI/ml, T 0.32 ng/ml, FAL 830 UI/L; ecografía: testículo derecho en canal inguinal e izquierdo ausente; DMO: osteoporosis L2-4 y cadera total, ambos con Z -2,8. Por síndrome de Kallmann se inició undecanoato de testosterona. Ya eugonádico se constató FAO 85.2 UI/l; centellograma óseo: aumento de fijación en articulaciones esternoclaviculares, hombros, calota, diáfisis y epífisis proximal de fémur izquierdo; Rx de calota: imagen en sal y pimienta. Por EP inició tratamiento con risedronato 35mg bisemanal, carbonato de calcio y ergocalciferol, constatándose a los 5 meses FAL 385 UI/l, FAO 16.3 UI/l, y a los 10 meses FAL 396 UI/l, FAO 13,7 UI/l. Luego de 18 meses normalizó la FAO, pero por discontinuar la consulta se desconoce la evolución de la DMO. Conclusiones: Se resalta la importancia de valorar la FAL en el seguimiento de varones hipogonádicos bajo TRH con aporte adecuado de calcio y vitamina D, ya que su elevación no solo reflejaría el impacto del tratamiento sobre el remodelado y mineralización ósea. Las dos patologías de estos casos clínicos son de muy baja prevalencia sin evidencia, a la fecha, de relación causal entre si. El seguimiento de la FAL/FAO permitió sospechar y diagnosticar la enfermedad de Paget en el contexto del hipogonadismo. FASEN 2012 Viernes 5 CASOS CLÍNICOS de octubre - Adrenal y Paratiroides - 37 y 38 - Salón Leguizamón - Coordinador: l gómez 159 Lasalle SCHWANNOMA ADRENAL DESCUBIERTO INCIDENTALMENTE Mana D, Lopardi M, Cazado E. Sección Endocrinología, Hospital de Agudos J.M. Penna. Pedro Chutro 3380, 1437 CABA, Argentina. daniela.mana@gmail.com 37 Introducción: El schwannoma es un raro tumor benigno originado en la vaina de mielina que recubre los nervios periféricos motores, sensitivos, sensoriales, simpáticos y de los pares craneales. Los schwannomas viscerales son extremadamente raros y en general son descubiertos incidentalmente. Originados en la glándula adrenal, han sido descriptos pocos casos. Caso clínico: Paciente mujer de 48 años que consulta derivada por cirugía para valoración hormonal por incidentaloma adrenal izquierdo de 7 cm. Refería haber comenzado hace un año con distensión abdominal y dispepsia, sin mejoría con tratamiento sintomático. Ante la persistencia de los síntomas se realizó una ecografía abdominal y se encontró una formación tumoral en suprarrenal izquierda. Como único antecedente de jerarquía la paciente era hipotiroidea bajo reemplazo con levotiroxina 50 µg/día. La ecografía abdominal describió una formación solida, lobulada, de 66 x 37 x 38 mm ubicada en el polo superior del riñón izquierdo. La tomografía computada (TC) mostró en la glándula suprarrenal izquierda una voluminosa formación hipodensa de contornos netos, con densidad de tejido blando y refuerzo heterogéneo post contraste, de 7 cm de diámetro. El laboratorio hormonal (Test de Nugent, SDHEA, testosterona, estradiol, catecolaminas urinarias, AVM, ARP/aldosterona) no presentó alteraciones. Se realizó cirugía laparoscópica sin complicaciones. La anatomía patológica informó: MACROSCOPIA: formación oval de 6,2 x 5,8 x 4 cm MICROSCOPIA: parénquima de glándula suprarrenal con histo-arquitectura conservada. Adyacente a la misma, sin infiltrarla, una proliferación delimitada de células de estirpe renal con núcleos elongados y citoplasma amplio que se disponen en haces cortos entrelazados. Acúmulos de histiocitos espumosos entremezclados. Vasos rodeados de tejido hialino. Cápsula delgada. Las técnicas de inmunohistoquímica arrojaron los siguientes resultados: -proteína S 100: positiva (CD34: negativo, Keratina 7: negativo, Actina músculo liso: negativo, Ki 67:1 %) Diagnóstico: adrenal normal. Schwannoma adrenal La paciente evolucionó en forma favorable y se encuentra asintomática. Discusión y conclusión: El schwannoma adrenal se origina en la médula adrenal a partir de nervios del plexo lumbar superior y del frénico o del vago. No se han descrito schwannomas originados en la corteza adrenal. Usualmente son benignos, de lento crecimiento, bien encapsulados, por lo que, en general, cuando son diagnosticados, presentan gran tamaño. No secretan hormonas, aunque se ha descripto un caso de schwannoma retroperitoneal productor de noradrenalina. Suelen ser asintomáticos aunque algunos pacientes refieren síntomas mínimos e inespecíficos como dolor abdominal o molestias dorsales. La imagen en la TC suele ser bien circunscripta, homogénea, redonda u oval. Degeneración quística y calcificaciones se describen en la etapa final de aquellos schwannomas de larga de evolución, lo cual podría sugerir su diagnóstico. El diagnóstico definitivo se realiza solo luego del examen anatomopatológico y la inmunotinción positiva para la proteína S100. Los schwannomas adrenales, aunque raros, deberían ser incluidos en el diagnóstico diferencial de tumores adrenales sólidos no funcionantes descubiertos incidentalmente. RAEM 2012. Vol 49 Nº Supl. 160 38 SÍNDROME DE CUSHING ECTÓPICO DE ORIGEN PANCREÁTICO Storani, ME, Subies FH, Bostico ST, Musich M. Hospital Municipal de San Isidro, Pcia. de Bs.As., Argentina. Introducción: El Síndrome de Cushing ectópico ocurre en el 5 al 10 % de los casos de hipercortisolismo ACTH dependiente. La localización más frecuente es la pulmonar. Presentamos un caso de Síndrome de Cushing ectópico de origen pancreático y de producción hormonal mixta. Material y método: Paciente sexo femenino de 46 años de edad, que consulta por aumento de 12 kilos de peso en un mes, poliuria, edema y mialgias en miembros inferiores, hipertricosis e híperpigmentación de cara, sudoración y polimenorrea. Examen Físico: rubicundez e híperpigmentación facial, distribución normal de la grasa, piel y TCS normal. TA: 140/100. Laboratorio: GB 12000(80 % neutrófilos), Na142, K4.9, TTOG: glucemia basal: 103, glucemia 120’: 179, calcemia: 9,1. Ante la sospecha clínica de hipercortisolismo se solicita: BASAL CORTISOL sérico 39/19ug/dl CLU779/139ug/24hs ACTH242/46pg/ml Post inhib 1mg dexa Post inhib 8mg dexa 42ug/dl 46,4ug/dl Estudios de imágenes: RX y TAC de tórax normal, RNM de hipófisis normal, Ecografía abdominal: Páncreas: imagen hipoecoica de 33mm en cuerpo, imagen quística bilobulada con septo de 44mm en cola. TAC y RNM de abdomen: en cabeza-cuello de páncreas formación expansiva de 24x27mm; en cuerpo, imagen seudo quística 42mm, dilatación del conducto de Wirsung. A nivel retroperitoneal 4 adenomegalias de 15 a 23mm y en parénquima hepático cuatro imágenes quísticas y sólidas en segmento II, IV y VIII. Glándulas suprarrenales normales sin lesiones focales. Barrido corporal con In111-octeotride: Tres focos de aumento de la captación del radiotrazador en región centroabdominal, que se fusionan con las lesiones descriptas a nivel del cuerpo-cabeza del páncreas y adenopatías lateroaórticas izquierdas y foco de híperfijación en el segmento VIII hepático. Estos hallazgos son compatibles con tejido que expresa receptores a somatostatina en la lesión primitiva pancreática, adenopatías e hígado. Evaluación hormonal neuroendocrina GLUCEMIA103 CEA 2,4/5ng/ml INSULINA 10/20uU/ml CATECOLAMINASNORMALES 5 hidroxiindolacètico 1,6 /6ng/24hs GH 0,20/7ng/ml CALCITONINA<5 GASTRINA 406/90pg/ml Evolución: Se indicó Ketoconazol 400mg/día con medición de cortisol 23ng/dl y mejoría del cuadro clínico. En plan de cirugía a realizarse a la brevedad con estudio de inmunohistoquímica de la anatomía patológica. Comentario: El objetivo de esta presentación es comunicar un caso de Síndrome de Cushing ectópico ACTH dependiente por su infrecuente localización y su escasa signosintomatología clínica en relación a la extensión tumoral. FASEN 2012 Viernes 5 de octubre CASOS CLÍNICOS - Gónada masculina, Hipófisis y Varios - 39 al 161 41 - Salón Castilla + Villalba - Coordinador: H Boquete HIPOFISITIS SECUNDARIA A IPILIMUMAB UNA ENTIDAD EMERGENTE QUE DEBEMOS CONOCER Aparicio MM, Bertini K, Ballarino MC, Mallea Gil S. 39 Hospital Militar Central. Servicio de Endocrinología. Av. Luis María Campos 726. 1425 Ciudad Autónoma de Buenos Aires. Argentina. Introducción: El ipilimumab es un anticuerpo monoclonal humano anti-CTLA-4 que bloquea el sitio de inhibición de la respuesta inmune en células T activadas y estimula la respuesta autoinmune antitumoral. Su uso está recientemente aprobado para melanoma avanzado y se encuentra en investigación para ser aplicado en múltiples cánceres. Su particular mecanismo de acción es la causa de variados efectos adversos de tipo autoinmune entre los cuales destacamos la hipofisitis. Caso clínico: Paciente masculino de 80 años con diagnóstico de adenocarcinoma de próstata con metástasis óseas de un año de evolución. Recibió tratamiento por tres meses con análogos de GNRH con respuesta parcial y posterior recaída y a continuación realizó tratamiento quimioterápico sin respuesta. Comienza esquema de inducción con ipilimumab como tratamiento de rescate. Recibe la primera infusión de 200 mg sin inconvenientes y al mes recibe la segunda. A los 10 días de la misma comienza con un cuadro de tipo gripal y dolor abdominal. A los 20 días es internado por deterioro del sensorio e hipotensión arterial asociados a astenia, adinamia, anorexia, constipación y empeoramiento del dolor abdominal. Clinicamente se presenta en regular estado general, hipotenso, afebril, con tendencia al sueño, con palidez de piel y mucosas, bradilálico, bradipsíquico, disfónico, con edema en miembros inferiores 2/6 y distensión abdominal. Laboratorio de ingreso: hto 26,5% (42-47), Hb 8.9 g/dl (13,8-15,8), Na 124 mmo/L (135-150), K 3,6 mmo/L (3,55,1), glucemia 1.55 g/lL (0,7-1,1), urea 1.06 g/L (0,1-0,5), creatinina 1,83 mg/dl (0,7-1,4), TSH 0.09 uU/ml (0,274,7), T4 3,63 ug/dl (4,5-12), FSH 1 mUI/ml (1,5-12,4), LH <0,1 mUI/ml (2-9), To 0,29 ng/ml (1,32-8,92), PRL 9 ng/ ml (4,6-21,4), IGF-1 31,5 ng/ml (94-267). Se interpreta el cuadro como hipopituitarismo secundario a hipofisitis autoinmune desencadenada por ipilimumab. Se indica reposición hidroelectrolítica, transfusión sanguínea, enema evacuante y 5 pulsos consecutivos de 1000 mg/día de metilprednisolona. Mejora la hiponatremia, la anemia y el estado general. Se indica 100 mcg/día de levotiroxina, 20 mcg/d de triiodotironina y continuar con 200 mg/d de hidrocortisona con lo que mejora aún más su estado general y laboratorio. Se realiza resonancia magnética nuclear con gadolinio de hipófisis que resulta normal. A los 10 días de la internación comienza con deposiciones diarreicas frecuentes que no responden al tratamiento con bismuto y loperamida. Se interpreta el cuadro como colitis autoinmune causada por ipilimumab. Reinicia metilprednisolona junto con 1 gr/d de mesalazina con escasa respuesta y gran deterioro de su estado general. A los 15 días manifiesta un exantema eritematopapuloso en rostro, tórax y miembros superiores interpretado como dermatitis autoinmune. Evoluciona con empeoramiento del estado general, gran deterioro del sensorio, hepatitis autoinmune, falla multiorgánica y finalmente óbito a los 20 días del ingreso. Conclusión: La hipofisitis secundaria a ipilimumab es una entidad de reciente aparición que los endocrinólogos debemos conocer. Los resultados alentadores que se observan en los pacientes que lo reciben hacen pensar que su uso más habitual y que será aplicada en múltiples cánceres, por lo tanto la hipofisitis se presentará con creciente frecuencia. El principal diagnóstico diferencial en este tipo de hipofisitis es el secundarismo del área selar y supraselar. El antecedente del tratamiento facilita su diagnóstico y tratamiento precoz pero aún así, el pronóstico del paciente depende de la presencia de otros efectos adversos presentes y de la enfermedad de base del paciente. RAEM 2012. Vol 49 Nº Supl. 162 40 TRATAMIENTO DE MACROADENOMA HIPOFISARIO PRODUCTOR DE HORMONA DE CRECIMIENTO CON DOSIS BIMENSUALES DE OCTREOTIDE Ferrada P, Cuello L, Ojeda A, Savina M, Torres E, Herrera J, Bringa J. Servicio de Enfermedades Endócrino – Metabólicas. Hospital Central de Mendoza. Argentina. Introducción: La cirugía transeptoesfenoidal continúa siendo el tratamiento de elección de los somatotropinomas. La misma debería ser realizada solo en centros con amplia experiencia (50 cirugías hipofisarias/ año). El tratamiento médico con análogos de somatostatina (octreotide, lanreotide), permite normalizar la secreción de GH en 2/3 de los pacientes y reducir el volumen tumoral en al menos 25 % casos. Las formulaciones de octeotride de acción prolongada se utilizan habitualmente cada 28 días. No está evaluada su eficacia al ser aplicadas en intervalos de tiempo mayores. Objetivo: Evaluar la eficacia de octeotride bimensual en un caso de adenoma quístico productor de GH. Caso clínico: Paciente de sexo femenino de 54 años de edad, quién consulta por cambios acros, artralgias y cefalea. Se solicita laboratorio: GH post TTOG basal, 60 y 120 min. 12, 30 y >40 ng/ml (VN hasta 1ng/ml) respectivamente, PRL 48.9 ng/ml, TSH 1.91 mU/L, T4libre 0.95 ng/dL, cortisol 23.9 μg/ml, LH 7.49 μU/ml, FSH 30.7 μU/ml, estradiol 9.49 pg/ml. RMN: macroadenoma quístico de 22 x 22 x 20mm (Imagen 1). Campo visual computarizado normal. Inicia tratamiento con octreotide LAR 20mg ampolla I.M. cada 60 días (por discontinuidad mensual debido a falta de medicación). En control de cuatro meses presenta desaparición de artralgias y cefalea con reducción del volumen tumoral de aprox. 50 % (13 x 18 x 17 mm, Imagen 2). Al año de tratamiento la paciente continua en tratamiento con octreotide bimensual, asintomática, normalización de GH e IGF-1 (0.7 ng/ml y 218 ng/ml, VR: 81-230 ng/ml, respectivamente) y resto tumoral de 6 x 8 x 7mm (reducción del aprox 70 % con respecto a lesión inicial, Imagen 3). Conclusión: Reportamos un caso de respuesta favorable a la administración bimensual de octreotide en el tratamiento de acromegalia producida por macroadenoma hipofisario. El tratamiento médico es una alternativa a la cirugía hipofisaria, que ha sido históricamente el único tratamiento propuesto a estos pacientes. Imagen 1 Imagen 2 Imagen 3 FASEN 2012 CASOS CLÍNICOS DIABETES MELLITUS, ACROMEGALIA E INSULINORRESISTENCIA SEVERA Ferrada P, Cuello L, Ojeda A, Savina M, Torres E, Juri A, Bringa J. Servicio de Enfermedades Endocrinometabólicas. Hospital Central de Mendoza. Argentina. 163 41 Introducción: la acromegalia, está incluida dentro de los tipos específicos de diabetes, siendo la insulinorresistencia hepática y periférica el mecanismo implicado. Ante el exceso de GH se describe una prevalencia de tolerancia anormal a la glucosa de 16 - 46 % y de diabetes mellitus y 19 - 56 %. Objetivo: presentar un caso de diabetes mellitus y acromegalia, con insulinorresistencia severa y dificultad para lograr un adecuado control glucémico. Caso clínico: mujer de 54 años de edad. Antecedentes: diabetes mellitus tipo 2 (10 años de diagnóstico, tratamiento: metformina 1700mg/día, insulina lispro 140 U/día. Diabetes gestacional en último embarazo a los 36 años y dos hermanos con diagnóstico de diabetes mellitus tipo 2), HTA (10 años de diagnóstico, tratamiento: enalapril, hidroclorotiazida), obesidad, tabaquismo no actual. Motivo de internación: tratamiento quirúrgico de macroadenoma hipofisario productor de GH con extensión supra e infraselar, invasión de seno cavernoso izquierdo y en contacto con quiasma óptico, diagnóstico realizado un año previo (paciente refiere síntomas de acromegalia desde 10 años antes), en tratamiento con lanreotide (90mg i.m.), habiendo recibido 3 dosis en dicho periodo de tiempo por dificultad en la continuidad del tratamiento. En seguimiento ambulatorio paciente presenta mal control glucémico a pesar de ajuste progresivo de insulinoterapia (glucemia capilar promedio de 350mg/dL, HbA1c 4 meses previos de 17 %). Al ingreso: peso 79Kg, talla 1,54m, IMC 33,3Kg/m2, perímetro cintura: 98cm, cambios acros, acrocordones, acantosis nigricans. Durante internación: clearance de creatinina 146 ml/min, relación albumina/creatinina en orina de 24hs 110,74 mg/gr, colesterol total 221mg/dL, HDL – colesterol 46mg/dL, triglicéridos 290mg/dL. Cortisol basal 13,9µg/ dL, PRL 4,9mg/mL, FSH 17,9mU/mL, TSH 0.9µU/mL, T4libre 0.9ng/dL, GH 21,1ng/mL, IGF-1 90.3ng/mL (VR 87 - 238, previo a internación ante última dosis de lanreotide). Fondo de ojo: retinopatía hipertensiva leve, ausencia de retinopatía diabética. Necesidad de aumento progresivo de dosis de insulina s.c. hasta 3.3U/Kg/día de insulina NPH y corriente. Paciente es sometida a cirugía de debulking, vía craneotomía. En la evolución posterior a tres meses de la cirugía: peso 82Kg, IMC: 34.4Kg/m2, clínicamente estable. Laboratorio: GH 19.9ng/mL, IGF-1 1004 ng/mL. Persistencia de mal control glucémico a pesar de 2.4U/Kg/día de insulina NPH (monitoreo glucémico 200 - 250mg/ dL). RMN selar: persistencia de resto de adenoma que ocupa región selar, pareselar izquierda con invasión del seno cavernoso y supraselar y parasagital izquierda en contacto con quiasma óptico y región hipotalámica. Discusión: la presentación de trastorno del metabolismo glucídico en acromegalia es frecuente, inclusive se describe insulinorresistencia en acromegálicos euglucémicos en comparación con población general. Factores de riesgo para desarrollo de diabetes mellitus en acromegalia: predisposición genética de DM, nivel de GH e IGF-1, edad avanzada, mayor duración de enfermedad. La elección del tratamiento del exceso de GH tiene también un impacto sobre el perfil glucémico, ya que la utilización de análogos de somatostatina puede disminuir la secreción de insulina, principalmente observado ante la utilización de octeotride – LAR. Ante el tratamiento quirúrgico se ha observado una mejoría de los niveles glucémicos y en la función de célula beta, independiente del valor de GH e IGF1 alcanzados. Esta situación es menos frecuente en pacientes con alteración significativa de la secreción de insulina pancreática prequirúrgica, como ocurre en nuestra paciente. En el paciente acromegálico, debido a la existencia de factores de riesgo cardiovascular (HTA, dislipidemia, obesidad) se observa un incremento de la morbimortalidad con respecto a la población general, de forma tal que sólo el 30 % de diabéticos al momento del diagnóstico de acromegalia sobreviven 20 años. Los trabajos se publicaron tal y como fueron enviados por los autores