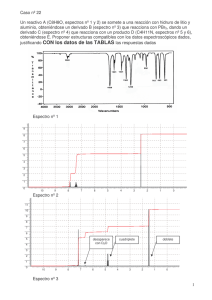
José Ramón Ramírez Díaz_Tesis_2013 - Ruidera
Anuncio

TESIS DOCTORAL “SÍNTESIS SOSTENIBLE DE NUEVOS DERIVADOS DE TRIAZINA. ESTUDIO DE SUS PROPIEDADES ÓPTICAS” Departamento de Química Inorgánica, Orgánica y Bioquímica UNIVERSIDAD DE CASTILLA-LA MANCHA FACULTAD DE CIENCIAS QUÍMICAS José Ramón Ramírez Díaz 2013 ÍNDICE Capítulo 1: Introducción general: química sostenible, radiación microondas y triazinas. 1. Introducción general…………………………………………………… 7 1.1.Química sostenible……………………………………………….…. 7 1.1.1. Desarrollo sostenible y química sostenible…………………… 9 1.1.2. Química sostenible y radiación microondas…………………. 10 1.2.La radiación microondas…………………………………………… 11 1.2.1. Calefacción microondas……………………………………… 12 1.2.1.1.Rotación dipolar………………………………………….. 1.2.1.2.Conducción iónica………………………………………… 1.2.2. Propiedades dieléctricas………………………………………. 14 1.2.3. Radiación microondas frente a calefacción clásica…………… 16 1.2.4. Equipamiento microondas…………………………………….. 19 1.2.4.1.Generador…………………………………………………. 1.2.4.2.Guía de onda………………………………………………. 1.2.4.3.Aplicador………………………………………………….. 1.2.4.3.1. Equipos microondas multimodo…………………… 1.2.4.3.2. Equipos microondas monomodo………………….. 1.2.5. Aplicación de microondas en síntesis química………………... 23 1.2.6. Efectos térmicos de la radiación microondas…………………. 24 1.2.7. Efectos no térmicos de la radiación microondas……………… 26 1.3. Importancia de las 1,3,5-triazinas…………………………………. 28 1.3.1. Fármacos……………………………………………………… 28 1.3.2. Herbicidas……………………………………………………... 30 1.3.3. Química supramolecular………………………………………. 31 1.3.3.1.Coordinación a metales…………………………………… 1.3.3.2.Enlaces de hidrógeno……………………………………… 1.3.3.3.Apilamiento………………………………………………. 1.3.4. Dispositivos optoelectrónicos…………………………………. 37 1.3.4.1.OLEDs…………………………………………………… 1.3.4.2.Óptica no lineal……………………………………………. 1.3.4.3.Células solares…………………………………………….. 1.3.5. Ciencia de los materiales……………………………………… 41 1.3.5.1.Cristales líquidos…………………………………………. 1.3.5.2.Catalizadores……………………………………………… 1.3.5.3.Nanotecnología……………………………………………. 1.3.5.4.Otras aplicaciones…………………………………………. Capítulo 2: Síntesis de nuevos derivados de 1,3,5-triazina con sistemas aromáticos. Estudio de sus propiedades ópticas. 2. Síntesis de nuevos derivados de 1,3,5-triazina con sistemas aromáticos. Estudio de sus propiedades ópticas……………………………………. 49 2.1. Objetivos……………………………………………………………. 49 2.2. Antecedentes bibliográficos……………………………………….. 50 2.3. Resultados obtenidos y discusión…………………………………. 57 2.3.1. Triazinas con 2,5-dimetoxianilina…………….………….….. 57 2.3.1.1. Síntesis………………………………………………….. 2.3.1.2. Determinación estructural………………………………. 2.3.1.3. Estudio de las propiedades ópticas…………………….. 2.3.1.3.1. Espectroscopia UV-visible……………………… 2.3.1.3.2. Fluorescencia……………………………………. 2.3.1.3.3. Excitación……………………………………….. 2.3.1.3.4. Influencia del disolvente……………………….. 2.3.1.3.5. Estudios de agregación…………………………. 2.3.2. Derivados de triazina con 1,5-diaminonaftaleno……………. 76 2.3.2.1. Síntesis…………………………………………………. 2.3.2.2. Determinación estructural……………………………… 2.3.2.3. Estudio de las propiedades ópticas……………………. 2.3.2.3.1. Espectroscopia UV-visible…………………….. 2.3.2.3.2. Fluorescencia………………………………….. 2.3.2.3.3. Excitación……………………………………… 2.3.2.3.4. Influencia del disolvente………………………. 2.3.2.3.5. Estudios de agregación………………………… 2.4. Parte experimental…………………………………………………. 101 2.4.1. Equipamiento………………………………………………… 101 2.4.2. Síntesis de monotriazinas con 2,5-dimetoxianilina………….. 104 2.4.3. Síntesis de derivados de triazina con 1,5-diaminonaftaleno…. 108 2.5. Conclusiones………………………………………………………... 113 Capítulo 3: Síntesis de nuevos derivados de triazinilglicina. Estudio de sus propiedades ópticas y en química supramolecular. 3. Síntesis de nuevos derivados de triazinilglicina. Estudio de sus propiedades ópticas y en química supramolecular………..………….. 117 3.1. Objetivos……………………………………………………………. 117 3.2. Introducción………………………………………………………... 119 3.2.1. Cationes metálicos…………………………………………… 119 3.2.1.1. Zinc……………………………………………………… 3.2.1.1.1. Fuentes dietéticas de zinc……………………….. 3.2.1.1.2. Ingesta dietética recomendada de zinc………….. 3.2.1.1.3. Déficit de zinc…………………………………… 3.2.1.1.4. Toxicidad del zinc………………………………. 3.2.1.2. Mercurio………………………………………………… 3.2.2. Sensores…………………………………………………….... 125 3.3. Antecedentes bibliográficos……………………………………….. 129 3.3.1. Detección de iones metálicos………………………………... 129 3.4. Discusión de resultados……………………………………………. 133 3.4.1. N,N’-bisaril-1,3,5-triazina-2,4,6-triaminas………………….. 133 3.4.1.1. Síntesis……………………………………………… 3.4.1.1.1. Ruta 1: Sintetizar derivados amino protegidos con un grupo lábil……. 3.4.1.2. Determinación estructural……………………………… 3.4.1.3. Ruta 2: Introducción del grupo amino en primer lugar… 3.4.2. N-triazinilglicinas…………………………………………… 140 3.4.2.1. Síntesis…………………………………………………. 3.4.2.2. Determinación estructural……………………………… 3.4.2.3. Estudios de 1H-RMN para el derivado de pirazol…….. 3.4.2.4. Estudios de difracción de Rayos-X………………….... 3.4.2.5. Estudio de las propiedades ópticas……………………. 3.4.2.6. Estudios con iones metálicos…………………………… 3.4.2.6.1. Sonda de mercurio………………………………. 3.4.2.6.2. Ensayos en células……………………………….. 3.4.2.6.3. Estudios con zinc………………………………… 3.4.3. Reacciones con nanoestructuras de carbono…………………. 177 3.5. Parte experimental…………………………………………………. 180 3.5.1. Equipamiento……………………………………………….... 180 3.5.2. Síntesis de aminotriazinas con 2,4-dimetoxibencilamina……. 180 3.5.3. Síntesis de 6-aminotriazinas-2,4-disustituidas……………….. 184 3.5.4. Síntesis general de derivados de N-triazinilglicina…………... 186 3.5.5. Experimentos de UV y fluorescencia……………………..…. 192 3.6. Conclusiones………………………………………………………... 195 4. Bibliografía……………………………………………………………… 199 5. Anexos………………………………………………………………….... 209 5.1. 12 principios de la Química Sostenible…………………………… 209 5.2. Espectros……………………………………………………………. 211 5.3. Tablas……………………………………………………………….. 281 1. Introducción general 7 1. INTRODUCCIÓN GENERAL. 1.1. QUÍMICA SOSTENIBLE. La química está viviendo una época difícil. Mientras la sociedad sigue demandando mayores cantidades de productos cada vez más sofisticados, también observa a las industrias que manufacturan estos productos con un grado creciente de sospecha y temor. El rango de productos químicos en la sociedad de hoy es enorme, y estos productos realizan una contribución a la calidad de nuestras vidas que no tiene precio. En medicina, el diseño y la manufactura de productos farmacéuticos nos han capacitado para curar enfermedades que han devastado a la humanidad a lo largo de la historia. Los productos de protección de las cosechas y los abonos nos han permitido incrementar nuestra producción de comida en gran medida. Es particularmente revelador mencionar que, aunque el siglo XX vio un crecimiento de la población mundial desde 1600 a 6000 millones de personas, también se ha visto un incremento en la esperanza de vida de aproximadamente un 60 %.1 La química ha jugado, y continúa jugando, un papel fundamental en casi todos los aspectos de la sociedad moderna y, como las enormes poblaciones de China, India y otras naciones emergentes demandan los niveles occidentales de asistencia sanitaria, comida, abrigo, transporte y bienes de consumo, las demandas de las industrias de productos químicos crecerán. El desarrollo satisfactorio de la industria química casi ha tenido una relación inversa con la percepción que la gente ha tenido de ella. Hay estudios que revelan que hace 10 años en Europa no había ninguna nación en la que la mayoría de la población tuviese una opinión favorable hacia la industria química. La interpretación más favorable de esos datos es que en algunos de los mayores núcleos de manufactura de productos químicos (como Alemania), hubo más gente que aportó puntos de vista positivos que negativos para la fabricación de productos químicos pero, para la mayoría 1 R. Breslau, Chemistry Today and Tomorrow. Awareness Chemical Society, Washington, 1997 1.1. Química sostenible 8 de países, la relación de opiniones desfavorables frente a las favorables era alarmantemente alta (Suecia, 2’8; Francia, 2’2; España, 1’5; Bélgica, 1’3). Debemos preguntarnos entonces por qué la química no tiene una buena imagen. La opinión pública es voluble y sujeta a la incomprensión y la confusión, reforzadas frecuentemente por los medios de comunicación. La industria farmacéutica, por ejemplo, está altamente considerada por el público a pesar del hecho de que representa una gran parte, que además va en aumento, de las industrias químicas. La “química” no causa la misma reacción hostil que los “productos químicos”, porque estos últimos son los que la gente asocia con los desastres, vertidos y aditivos no deseados a sus alimentos, bebidas o bienes de consumo. Es revelador notificar un cambio en el nombre de la asociación comercial de las industrias químicas líder en EEUU, desde Asociación Americana de Fabricantes de Productos Químicos hasta Consejo Americano de la Química. En efecto, un punto de vista cínico podría ser que podemos resolver nuestros problemas de la noche a la mañana reinventándonos como “diseñadores moleculares”.2 ¿Qué necesitamos para cambiar esta mala imagen? Aunque los primeros trabajos para fabricar los productos químicos de una manera más sostenible iban encaminados mayoritariamente a reducir el impacto que los procesos tenían sobre el medio, en el futuro será necesaria una visión más amplia. De forma exagerada, podemos escribir una receta con los pasos que se seguían en el siglo pasado para realizar un proceso químico: a) Parta de una materia prima basada en el petróleo. b) Disuélvala en un disolvente. c) Añada un reactivo. d) Reacción para formar un intermedio químico. e) Repita los pasos del 2 al 4 hasta obtener el producto final; deseche todos los residuos y reactivos gastados; recicle el disolvente cuando sea viable económicamente. f) Transporte el producto por todo el mundo, frecuentemente para un almacenamiento prolongado. g) Libere el producto en el ecosistema sin evaluar sus efectos a largo plazo. 2 J. Clark, D. Macquarrie, Handbook of Green Chemistry & Technology, Blackwell Science, 2008 1. Introducción general 9 El procedimiento para el futuro debe ser muy diferente: a) Diseñe la molécula para que tenga el mínimo impacto en el medio ambiente (tiempo de residencia bajo, biodegradable) b) Fabrique a partir de una materia prima renovable (por ejemplo, carbohidratos). c) Use un catalizador de vida larga. d) No use disolvente o, en caso de usarlo, que sea benigno y reciclable totalmente. e) Use el menor número de pasos posible en la síntesis. f) Fabrique el producto cuando se requiera y lo más cerca posible de donde se requiera. El escenario se debe aplicar no sólo en química, sino también en transporte, en legislación y, de forma más crítica, en la educación. Las nuevas generaciones de químicos deben estar entrenadas para pensar en los factores medioambientales, sociales y económicos de los procesos de fabricación de productos químicos. 1.1.1. DESARROLLO SOSTENIBLE Y QUÍMICA SOSTENIBLE En el contexto moderno, los términos “desarrollo sostenible” y “química sostenible” han existido durante los últimos 25 años. La discusión sobre la sostenibilidad empezó, esencialmente, cuando en 1987 la Comisión de Desarrollo y Medio Ambiente de la ONU (conocida como la Comisión Bruntland) notificó que el desarrollo económico podría conducir a un deterioro, y no a una mejora, de la calidad de vida de la población.3 Esto nos llevó hasta la comúnmente aceptada definición de “desarrollo sostenible” como: “Desarrollo que afronta las necesidades del presente sin comprometer la capacidad de las generaciones futuras de afrontar sus necesidades”. 3 World Commision of Environment and Development. Our Common Future. Oxford University Press. Oxford. 1987 1.1. Química sostenible 10 Esta definición es intencionadamente muy amplia, cubriendo todos los aspectos de la sociedad. El desarrollo sostenible tiene una particular relevancia en las industrias basadas en la química porque está interesado en evitar la contaminación y el uso temerario de los recursos naturales. En esencia, se está reconociendo como la búsqueda de los principios y metas de la química sostenible. El movimiento de la química sostenible empezó en los primeros años de la década de 1990 promovido por la Agencia de Protección Medioambiental de Estados Unidos (EPA)4 como un medio para incentivar a la industria y al mundo académico a usar la química como prevención de la contaminación. Más específicamente, la misión de la química sostenible era: “Promocionar tecnologías químicas innovadoras que reducen o eliminan el uso o generación de sustancias químicas peligrosas en el diseño, manufactura y uso de productos químicos”. En conjunción con la Sociedad Americana de Química (ACS),5 la EPA desarrolló la química sostenible en 12 principios,6 que se pueden ver en el anexo I, en los que se puede resumir el espíritu de este concepto. 1.1.2. QUÍMICA SOSTENIBLE MICROONDAS Y RADIACIÓN Las técnicas de síntesis no convencionales pueden ser consideradas como métodos sostenibles. Dentro de éstas cabe destacar la síntesis asistida por microondas. Esta técnica repercute positivamente, sobre todo, en el sexto principio de la química sostenible (disminuir el consumo energético). Indirectamente también influye en otros principios, como la reducción y el uso de sustancias auxiliares (sobre todo en las reacciones en las que combinamos la radiación microondas con la ausencia de disolvente). Environmental Protection Agency. American Chemical Society. 6 P. T. Anastas, J. C. Warner. Green Chemistry. Theory and Practice. Oxford University Press. 1998. 4 5 1. Introducción general 11 1.2. LA RADIACIÓN MICROONDAS La radiación microondas7 es una alternativa a la calefacción clásica como método de introducir energía en las reacciones químicas, aprovechando la capacidad de algunos compuestos de transformar la energía electromagnética en calor. Identificamos como radiación microondas a las ondas electromagnéticas comprendidas en el espectro entre longitudes de onda de 1 cm a 1 m, correspondiendo con frecuencias desde 30 GHz a 300 MHz (figura 1.1). Debido a que los radares y otros sistemas de telecomunicaciones utilizan frecuencias ubicadas en la zona de las microondas, para evitar interferencias existen acuerdos internacionales que regulan el uso de estas frecuencias. Las frecuencias de uso industrial, científico y médico (ISM) están reguladas en 5’8 GHz, 2’45 GHz (12’2 cm) y 900 MHz (33’3 cm) para la calefacción dieléctrica microondas. Los hornos domésticos y los reactores microondas con fines químicos generalmente operan a 2’45 GHz, por lo que la mayor parte de la literatura sobre este tema está basada en equipos que trabajan a esta frecuencia. Figura 1.1. Espectro electromagnético. Podemos relacionar energía (E), longitud de onda (λ) y frecuencia (ν) de la radiación mediante la siguiente ecuación, donde c es la velocidad de la luz: E=h·ν=h·c/λ 7 Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012. 12 1.2. La radiación microondas La energía de un fotón de microondas en esta región de frecuencias del espectro es de 0’0016 eV, la cual es muy baja para romper enlaces químicos. Por lo tanto, las microondas no pueden “inducir” reacciones químicas por absorción directa de energía electromagnética, al contrario de lo que ocurre con la radiación visible y ultravioleta.8 El uso del microondas está permitiendo reescribir muchas reacciones clásicas con un gran ahorro térmico y mejora del rendimiento. Debido a sus beneficios medioambientales, la síntesis asistida por microondas es un método que está cobrando importancia últimamente. Estos hechos se ponen de manifiesto con el aumento de publicaciones en estos últimos años, acompañado con la aparición de una instrumentación más moderna.9 1.2.1. CALEFACCIÓN MICROONDAS El uso del microondas como forma de calefacción tiene numerosos atractivos en química debido a que, al contrario que la calefacción clásica, la velocidad de calefacción depende de la naturaleza de las moléculas, en particular de las propiedades dieléctricas, por lo que puede considerarse como una calefacción selectiva. Las microondas son ondas electromagnéticas compuestas por una componente eléctrica y otra magnética. En la mayor parte de los casos es la componente eléctrica de la radiación microondas la principal responsable de la interacción onda-materia, aunque en algunos casos la interacción magnética puede ser relevante, como en presencia de óxidos de metales de transición.7 Existen dos mecanismos principales para llevar a cabo la calefacción con microondas: rotación dipolar y conducción iónica. Ambos mecanismos requieren un a) D. Stuerga, Microwaves in Organic Synthesis, Ed.: A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012. b) M. D. P. Mingos Microwave-Asssisted Organic Synthesis, Eds.: P. Lidström, J. P. Tierney, Blackwell, Oxford, 2005. 9 a) A. de la Hoz, A. Díaz-Ortiz, A. Moreno, Curr. Org. Chem., 2004, 8, 903. b) C. O. Kappe, Angew. Chem. Int. Ed., 2004, 43, 6250. c) M. Nüchter, B. Ondrushka, W. Bonrath, A. Gum, Green Chem., 2004, 6, 128. d) D. Bogdal, A. Loupy, Org. Proc. Res. & Dev., 2008, 12, 710. e) C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127. f) V. Polshettiwar, R. Varma, Chem. Soc. Rev., 2008, 37, 1546. g) C. O. Kappe, A. Stadler, D. Dallinger, Microwaves in Organic and Medicinal Chemistry, 2nd Ed. Wiley-VCH, Weinheim, 2012. 7 Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012, pág. 30. 8 1. Introducción general 13 acoplamiento efectivo entre los componentes del material y la oscilación rápida de la componente eléctrica de la radiación microondas. 1.2.1.1. Rotación dipolar: Las moléculas con momento dipolar permanente o inducido, bajo la influencia de un campo eléctrico externo, tienden a alinear su momento dipolar con éste (figura 1.2). A la frecuencia de microondas, el campo cambia de dirección unas 4’9 x 109 veces por segundo, frecuencia que se sitúa entre los extremos anteriormente citados. Como el campo aplicado oscila, los dipolos tienden a alinearse con éste dando tiempo a los dipolos para alinearse con el campo, pero no para seguir su alternancia. Dipolos en ausencia de campo Dipolos en presencia de campo eléctrico eléctrico Figura 1.2. Dipolos en disolución sin y con campo eléctrico. Así, se genera una diferencia de fase entre el dipolo y la orientación del campo. Esta diferencia de fase causa que la energía se pierda en forma de calor por fricciones y colisiones moleculares y por pérdidas dieléctricas, dando lugar a la calefacción dieléctrica por el mecanismo de rotación dipolar. La cantidad de calor generada es directamente proporcional a la capacidad de los dipolos de alinearse con el campo aplicado. Si los dipolos no tienen tiempo de alinearse o se alinean demasiado rápido, la muestra no se calienta. Los gases no pueden calentarse mediante irradiación microondas, debido a que las distancias entre las moléculas en rotación son demasiado grandes, aparte de no poseer momentos dipolares adecuados para las pérdidas dieléctricas. Además, los sólidos cristalinos son prácticamente transparentes a la radiación microondas, debido a la escasa movilidad de las moléculas que forman la red cristalina, en contraste con la movilidad de las moléculas en el seno de un líquido. 14 1.2. La radiación microondas 1.2.1.2. Conducción iónica: Otro mecanismo importante de calefacción con microondas es la conducción iónica. Durante la aplicación del campo magnético oscilante, las partículas cargadas disueltas (iones) en una muestra oscilan de un lado a otro, provocando colisiones con los átomos o moléculas vecinas. Estas colisiones causan agitación o movimiento, produciendo calor. De este modo, si calentamos dos muestras que contienen la misma cantidad de agua desionizada y agua mineral, respectivamente, con radiación microondas a una potencia y a un tiempo fijo, el agua mineral se calentará más rápido debido a su contenido en iones. Los efectos de la conducción iónica son especialmente importantes cuando se trabaja con líquidos iónicos en microondas. En este caso, el mecanismo de conducción iónica es mucho más fuerte que el de rotación dipolar en cuanto a lo que a generación de calor se refiere. Asimismo, el mecanismo de conducción iónica permite explicar el hecho de que algunos metales cuyo momento dipolar es nulo, se calientan efectivamente con microondas. 1.2.2. PROPIEDADES DIELÉCTRICAS Es útil cuantificar la capacidad de transformar la radiación microondas en calor. Esta magnitud física se denomina factor de disipación (tan δ). Valores altos de este parámetro indican una fácil susceptibilidad a la radiación microondas. Este factor se define como el cociente entre la pérdida dieléctrica (ε’’), que indica la eficiencia con la que la radiación electromagnética se transforma en calor y la constante dieléctrica (ε’), que describe la polarizabilidad de la molécula en un campo eléctrico. Además, esta relación (ε’’/ε’) describe la diferencia de fase entre el campo eléctrico y la polarización del material. La frecuencia y la temperatura son parámetros que influyen en el factor de disipación y en la polarización del material. En la tabla 1.1 se muestran algunas 1. Introducción general 15 sustancias con sus correspondientes valores del factor de disipación (con ν = 3 GHz y T = 298 K): Tabla 1.1. Factor de disipación (tan δ) de diferentes sustancias. Material ε’ ε’’ tan δ x 104 Hielo 32’7 0’0288 9 Agua 76’7 12’0419 1570 NaCl 0’1 M 75’5 18’12 2400 Metanol 23’9 15’296 6400 Etanol 6’5 1’625 2500 CCl4 2’2 0’00088 4 Heptano 1’9 0’00019 1 Otro factor importante para describir el proceso de calefacción es la penetración de la onda en el material (PD)10. Se considera por convenio como penetración de la onda a la profundidad del sistema a la cual la onda disminuye su intensidad a un 37 % de su valor inicial. La frecuencia a la que operan los reactores microondas comerciales (2450 MHz) proporciona una penetración de la onda de únicamente unos pocos centímetros, dependiendo de las propiedades dieléctricas específicas del sistema, según la siguiente ecuación: √′ ′′ Una mayor frecuencia implica una mayor energía, pero una menor longitud de onda. Teniendo en cuenta este razonamiento, y viendo la ecuación anterior, podemos deducir que si una mayor frecuencia implica una mayor energía, pero una menor longitud de onda, disminuye la penetración de la misma, lo que llevaría a un calentamiento superficial y no de todo el volumen de la muestra, cosa que no es interesante. Por tanto, debemos llegar a un compromiso entre la energía de la radiación y la penetración de la misma en el material. En la tabla 1.2 se muestran algunos valores de PD para diferentes sustancias: 10 Penetration Depth. 16 1.2. La radiación microondas Tabla 1.2. Factor de penetración (PD) para diferentes sustancias. Sustancia PD (2’45 GHz) Agua (25 ºC) 1’4 cm Agua (95 ºC) 5’7 cm Hielo (-12 ºC) 11 m Cuarzo (25 ºC) 160 m Teflón (25 ºC) 9’2 m Sustancias con un factor de penetración muy grande, como el cuarzo son prácticamente transparentes a la radiación microondas. 1.2.3. RADIACIÓN MICROONDAS FRENTE A CALEFACCIÓN CLÁSICA Tradicionalmente, la síntesis orgánica se ha llevado a cabo mediante calefacción por conducción mediante una fuente externa de calor. Existen diferencias apreciables entre la calefacción dieléctrica con microondas y la calefacción clásica. A continuación expondremos algunas de las más importantes: Una de las grandes ventajas de la irradiación con microondas es que la distribución de la temperatura en el volumen de la muestra es más homogéneo que en calefacción clásica, como puede verse en la figura 1.3, en la cual se muestra claramente que en la calefacción clásica (a la izquierda) se calienta en primer lugar la superficie del recipiente que contiene la mezcla de reacción, transmitiéndose el calor por conducción y convección, mientras que en la calefacción por microondas se calienta el contenido del matraz en primer término, ocurriendo la transmisión del calor mediante pérdidas dieléctricas. 1. Introducción general 17 La temperatura en la superficie es mucho mayor. El recipiente no se calienta. La temperatura en el interior es mayor. La energía se transmite por pérdidas dieléctricas. La energía se transmite por corrientes de convección. Calefacción conductiva Calefacción con microondas Figura 1.3. Diferencias de mecanismo entre calefacción clásica y microondas. Las velocidades de calefacción que proporciona la irradiación con microondas pueden ser mucho mayores que las obtenidas mediante calefacción clásica, y a menudo no pueden ser reproducibles con este tipo de calefacción. Este hecho permite una notable reducción de los tiempos de reacción, lo que evita la descomposición térmica de los productos o reactivos sensibles. La figura 1.4 es ilustrativa acerca de la diferencia en el perfil térmico de las reacciones activadas mediante calefacción clásica y las reacciones activadas por microondas tras un minuto de calentamiento: Figura 1.4. Perfiles de temperatura tras un minuto de reacción. Este efecto es particularmente importante en: 18 1.2. La radiación microondas • La preparación de fármacos marcados con isótopos que tienen una vida media corta,11 como por ejemplo: 122I, t1/2 = 36 min. • Química combinatoria, ya que permite la preparación de un mayor número de productos en un tiempo más corto.12 • Catálisis,13 donde tiempos de reacción cortos pueden evitar la descomposición del catalizador y aumentar su eficacia. La calefacción dieléctrica con microondas es más selectiva que la calefacción convencional, ya que depende de las propiedades del material. Compuestos polares se calientan muy eficazmente, mientras que los compuestos apolares no se calientan apreciablemente. Por el contrario en calefacción convencional la calefacción depende casi exclusivamente de la temperatura que fijemos en el sistema de calefacción. En la tabla 1.3 podemos ver la diferencia entre la velocidad de la calefacción convencional, con un baño de silicona, y la calefacción con microondas, comparando las temperaturas que alcanzan una serie de disolventes tras un minuto de calentamiento. Tabla 1.3. Temperaturas que alcanzan distintos disolventes tras un minuto de calefacción. T (ºC) 1 min Disolvente T Eb (ºC) Baño MO Agua 39 81 100 CCl4 38 28 77 DMF 43 131 153 Etanol 66 78 78 El proceso de ebullición de los disolventes es tanto termodinámico como cinético. Los disolventes pueden sobrecalentarse con microondas por encima de su punto de ebullición antes de que se produzca la misma.14 De esta manera pueden conseguirse condiciones no reproducibles mediante calefacción convencional. N. Elander, J.R. Jones, S. Y. Lu and S. Stone-Elander, Chem. Soc. Rev, 2000, 29, 239. H. E. Blackwell, Org. Biomol. Chem., 2003, 1, 1251. 13 N. F. Kaiser, U. Bremberg, M. Larhed, C. Moberg and A. Hallberg. Angew. Chem. Int. Ed, 2000, 39, 3595. 14 D. R. Baghurst, D. M. P. Mingos, J. Chem Soc., Chem. Commun., 1992, 674. 11 12 1. Introducción general 19 En muestras sólidas, la transferencia de energía es más lenta, por lo que es más fácil la existencia de puntos calientes (véase apartado 1.2.6).7 Analizando el consumo de energía de las reacciones, se ve que el balance energético es favorable a las reacciones inducidas por radiación microondas, en comparación con la manta calefactora o el baño de aceite, que es el menos eficaz en este sentido, como puede verse en la figura 1.5. Este hecho es especialmente relevante si tenemos en cuenta que nos encontramos en una época de escasez de recursos y crisis económica. Figura 1.5. Comparativa de consumo energético de diferentes sistemas de calefacción. 1.2.4. EQUIPAMIENTO MICROONDAS Los reactores microondas diseñados para síntesis química son similares a cualquier otro equipo de microondas. Sus partes básicas son un generador de microondas, un espacio físico donde la muestra absorbe la radiación microondas (aplicador) y un sistema de conducción de la radiación microondas desde el generador hasta el aplicador, llamado guía de onda. Para controlar las reacciones, también es necesario un sistema de monitorización que controle potencia, temperatura y presión en el reactor. 1.2.4.1. Generador: El generador es el componente que se encarga de suministrar a las muestras la energía electromagnética. Existen numerosos tipos de generadores, si bien el más usado 7 Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012, pág. 43, 249-256. 20 1.2. La radiación microondas es el magnetrón. Consiste en un sistema de alto vacío donde un cátodo separado de un ánodo es calentado por un alto voltaje, alrededor de 4 kV, en presencia de un fuerte campo magnético axial. El ánodo consta de un número par de cavidades, normalmente ocho, cada una de las cuales se comporta como un circuito regulado con un final abierto como una capacitancia. Cada cavidad actúa como un oscilador eléctrico que resuena a una frecuencia específica. Los electrones emitidos por el cátodo se dirigen hacia el ánodo acelerados por la diferencia de potencial entre ambos. La trayectoria que siguen estos electrones hacia el ánodo es en forma de espirales, debido a la presencia del fuerte campo magnético. En la figura 1.6 podemos ver una representación de un magnetrón: Figura 1.6. Esquema de un magnetrón. La eficiencia de un magnetrón es del 60 % o menor. La pérdida de rendimiento se produce en forma de calor, por lo que es necesario un sistema de refrigeración por aire frío que evite un excesivo calentamiento. 1.2.4.2. Guía de onda: Es el canal responsable de transportar las ondas desde el generador hasta el aplicador. Sus dimensiones están ligadas a la frecuencia que transporta. 1.2.4.3. Aplicador: Es un sistema diseñado para asegurar la transferencia de energía desde el magnetrón hasta la muestra. Existen dos tipos de aplicadores: multimodo y monomodo. 1. Introducción general 1.2.4.3.1. 21 Equipos microondas multimodo: El horno microondas consiste en un generador de microondas, circuitos de control de potencia y una cavidad para la muestra. La cavidad es de acero inoxidable que refleja la radiación, que no sólo evita que escape la radiación fuera del horno sino que cuando las microondas entran en la cavidad se reflejan repetidamente sobre la muestra donde son absorbidas en cada paso. En general la cavidad es muy grande en relación a la muestra y a la longitud de onda de la radiación. Con o sin muestra dentro del horno, las microondas que entran dentro de la cavidad se reflejan en las paredes dando una forma compleja de ondas estáticas. Este hecho es un problema importante cuando se usan hornos domésticos. Sin embargo, en sistemas multimodo diseñados para síntesis química se emplean dos magnetrones y una pirámide difusora, que minimiza la heterogeneidad del campo. Estos sistemas multimodo son sistemas pulsados, es decir, trabajan siempre a la máxima potencia y el control del sistema se lleva a cabo conectando y desconectando el magnetrón. En la figura 1.7 se muestra un esquema de un equipo microondas multimodo: Figura 1.7. Esquema de un equipo microondas multimodo. 1.2.4.3.2. Equipos microondas monomodo o focalizados: En un reactor monomodo la radiación se enfoca a la muestra a través de una guía de ondas, que tiene una anchura del orden de la longitud de onda, lo que permite enfocar la onda al material, aumentando la eficacia de la radiación. Por otra parte, la posibilidad de controlar la potencia de irradiación y la temperatura, haciendo que ésta 22 1.2. La radiación microondas sea constante, y el hecho de que la distribución de la energía sea uniforme, hace que los resultados sean más reproducibles. En el horno monomodo la cavidad es muy reducida, adaptada al matraz donde se coloca la muestra, y la radiación está focalizada hacia la cavidad lo que no solo hace que la radiación sea más intensa sino que también sea más homogénea en toda la cavidad. Asimismo se trata de un sistema abierto, lo que permite adaptar equipamiento de laboratorio convencional. A continuación se muestra un esquema de una cavidad monomodo (figura 1.8): Figura 1.8. Esquema de un equipo microondas monomodo. En la tabla 1.4 mostramos a modo de resumen las diferencias entre los equipos multimodo y monomodo. La elección de un modelo u otro depende de la aplicación deseada. Tabla 1.4. Multimodo vs Monomodo. Cavidad multimodo Cavidad monomodo Cavidad grande Cavidad compacta Trabajo a gran escala (5-1000 mL) Trabajo a pequeña escala (0’5-50 mL) Facilidad de escalado Escalado por flujo continuo Síntesis paralela Eficacia por automatización Campo no homogéneo Campo altamente homogéneo Baja densidad de potencia Alta densidad de potencia Alta potencia de salida Menor potencia de salida Problemas a pequeña escala Gran escala necesita tiempos largos 1. Introducción general 1.2.5. 23 APLICACIÓN DE MICROONDAS EN SÍNTESIS QUÍMICA A pesar de que la aplicación de las microondas en química orgánica surgió años después que en otras áreas de la química, el uso de las microondas en esta área de la química emergió de forma exitosa y con un desarrollo vertiginoso desde 1986,15 siendo actualmente el área donde se producen resultados más relevantes empleando esta tecnología. Se ha visto que la calefacción mediante radiación microondas otorga algunas ventajas importantes sobre la calefacción clásica, lo que la hace muy útil en numerosas reacciones de química orgánica. La síntesis orgánica asistida por microondas (MAOS – Microwave Assisted Organic Synthesis-) presenta unas ventajas positivas desde el punto de vista de la química sostenible, como: o Aceleración de reacciones y disminución de los tiempos de reacción. Las reacciones llevadas a cabo bajo radiación microondas tienen lugar en un tiempo menor que las que se realizan en las mismas condiciones bajo calefacción clásica. Esta notable reducción de tiempos de reacción evita la descomposición térmica de productos o reactivos sensibles y permite la obtención de rendimientos más altos en condiciones de reacción más suaves. o Mejora de los rendimientos de reacción y disminución de los productos secundarios. Las reacciones bajo radiación microondas suelen llevarse a cabo a temperaturas relativamente altas, comparando con experimentos similares realizados bajo calefacción clásica. Este hecho implica varias consecuencias, además de un aumento de velocidad de reacción. Las reacciones bajo microondas se llevan a cabo en condiciones controladas y optimizadas, lo que hace que el mecanismo de reacción esté bastante controlado. Esto conlleva la formación de menos productos secundarios, lo que hace las a) R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge, J. Rousell, Tetrahedron Let.., 1986, 27, 279. b) R. J. Giguere, T. L. Bray, S. M. Duncan, G. Majetich, Tetrahedron Let.., 1986, 27, 4945. 15 24 1.2. La radiación microondas reacciones más limpias. Además, los rendimientos de reacción en algunos casos sufren aumentos espectaculares. o Reacciones que sólo tienen lugar bajo irradiación microondas. Existen numerosos ejemplos en la literatura en los que la radiación microondas modifica la quimio y regioselectividad de las reacciones.7 1.2.6. EFECTOS TÉRMICOS DE LA RADIACIÓN MICROONDAS Los efectos térmicos observados bajo irradiación microondas son consecuencia de una transferencia de calor inversa, las inhomogeneidades del campo microondas en las muestras y una absorción selectiva de la radiación por parte de las moléculas polares. Estos efectos pueden usarse para hacer los procesos más eficientes o para modificar la selectividad de las reacciones. Sobrecalentamiento: Podemos explotar en la práctica el sobrecalentamiento de compuestos polares. Este efecto fue determinado por Mingos en líquidos polares bajo irradiación microondas (figura 1.9).14 El sobrecalentamiento se situaba entre 13 y 26 ºC sobre la temperatura de ebullición, y se debe principalmente al incremento de absorción de la radiación conforme aumenta la temperatura. También la transferencia de calor inversa contribuye a este sobrecalentamiento. Este efecto puede explicar el incremento en la velocidad de las reacciones orgánicas y organometálicas, es difícilmente reproducible mediante calefacción clásica y se puede usar para mejorar la eficiencia y los rendimientos de algunos procesos. 7 Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012. D. R. Baghurst, D. M. P. Mingos, J. Chem Soc., Chem. Commun., 1992, 674. 14 1. Introducción general 25 Etanol Figura 1.9. Sobrecalentamiento observado para etanol. Puntos calientes: Varios autores han postulado la existencia de puntos calientes en muestras irradiadas con microondas. Este efecto térmico procede de la inhomogeneidad del campo electromagnético aplicado, y tiene como resultado que la temperatura en determinadas zonas sea mucho mayor que la temperatura macroscópica de la muestra. Así, las condiciones de reacción globales (macroscópicas) no son representativas de las condiciones de reacción (microscópicas), como muestra la figura 1.10. 7 P3 a) P1 b) P2 P2 P1 P3 Figura 1.10. a) Fotografía de la superficie de una mezcla de reacción, llevada a cabo bajo radiación microondas. b) Termovisión de la superficie de la mezcla de reacción. 7 Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012, págs. 249256. 26 1.2. La radiación microondas Calefacción selectiva: Las propiedades dieléctricas de las microondas pueden proporcionar una calefacción selectiva de unos componentes de la reacción en presencia de otros. Cuando la velocidad de reacción es lo suficientemente rápida para que la transferencia de calor por mecanismos convencionales sea casi irrelevante en el futuro de la reacción, pueden observarse diferencias apreciables entre la calefacción clásica y la calefacción bajo radiación microondas. Esta calefacción puede usarse para calentar selectivamente disolventes, algún reactivo o el catalizador. 1.2.7. EFECTOS NO TÉRMICOS DE LA RADIACIÓN MICROONDAS Como hemos dicho anteriormente, la radiación microondas es capaz de mejorar el rendimiento e incluso modificar la selectividad de algunas reacciones. Hay autores que postulan la existencia de efectos no térmicos, denominados efecto microondas,16 debidos a la capacidad altamente polarizante del campo que, junto con su influencia en procesos de movilidad y difusión de las moléculas, hace que aumente la probabilidad de choques efectivos. Estos efectos no térmicos también provienen de las interacciones del campo electromagnético y el material, de forma similar a lo que ocurre con los efectos térmicos. Como consecuencia, ambos efectos aparecerán de forma simultánea y no es fácil separarlos. Mientras que los efectos térmicos están ampliamente aceptados y probados, los efectos no térmicos son objeto de gran controversia entre la comunidad científica. Existen trabajos que defienden su existencia y otros, entre los que destacan los de Kappe, que la rechazan, resaltando la importancia de un buen sistema de agitación y precisión en la medida de la temperatura a la hora de razonar los efectos de la radiación microondas.17 a) A. de la Hoz, A. Díaz-Ortiz, A. Moreno, Chem. Soc. Rev., 2005, 34, 164. b) A. de la Hoz, A. Díaz-Ortiz, A. Moreno, J. Microwave Power Electromagn. Energy, 2007, 41, 44. c) B. Pchelka, A. Loupy, A. Petit, Tetrahedron, 2006, 62, 10968. 17 M. A. Herrero, J. M. Kremsner, C. O. Kappe, J. Org. Chem., 2008, 73, 36. 16 1. Introducción general 27 Uno de los últimos trabajos de Kappe tenía como objetivo separar los efectos térmicos de los no térmicos en la radiación microondas18. Su idea consistió en realizar diferentes reacciones en matraces de SiC (que absorben muy eficazmente la radiación microondas) y en matraces de vidrio Pyrex (transparentes a la radiación microondas). De esta forma, en los matraces de SiC sólo tendremos efectos térmicos de la radiación, mientras que en los de vidrio Pyrex se observarían tanto los efectos térmicos como los no térmicos. Se vio que los resultados en ambos matraces eran comparables, de lo que se deduce que en las reacciones que probó Kappe no se observan efectos no térmicos. Ahora bien, un detalle importante de estas reacciones es que todas se realizaron a alta temperatura y, en estas condiciones, los efectos térmicos son mucho más importantes que los efectos no térmicos. Los efectos del tipo de disolvente, volumen, material del recipiente, velocidad de agitación en la distribución del campo eléctrico, densidad de la potencia y velocidad de calentamiento son una serie de fenómenos que deberían considerarse antes de diseñar unas conclusiones sobre efectos térmicos y no térmicos. De hecho, hay numerosas situaciones en las que las observaciones experimentales son frecuentemente malinterpretadas como “efecto microondas”. 18 D. Obermayer, B. Gutmann, C. O. Kappe, Angew. Chem. Int. Ed., 2009, 48, 44, 8321 28 1.3. Importancia de las 1,3,5-triazinas 1.3. IMPORTANCIA DE LAS 1,3,5-TRIAZINAS: Los compuestos derivados de 1,3,5-triazina (figura 1.11) han despertado un gran interés para la comunidad científica, debido a sus interesantes propiedades.19 Así, estos derivados han encontrado aplicaciones en múltiples campos, describiéndose a continuación algunas de las más importantes. Figura 1.11. Anillo de 1,3,5-triazina. 1.3.1. FÁRMACOS Si nos fijamos en la estructura del anillo de 1,3,5-triazina, observamos bastante similitud con la que presentan compuestos tan importantes para la vida como son las bases nitrogenadas, en especial las bases pirimidínicas (figura 1.12), que forman parte de los nucleótidos, que son el monómero constituyente de ADN y ARN. Figura 1.12. Bases pirimidínicas. Viendo este hecho, no nos debería extrañar que se haya observado actividad citotóxica en algunos derivados de 1,3,5-triazina,20 que puede aprovecharse para el tratamiento de enfermedades, como el cáncer. En esta línea, Peterson y colaboradores21 han logrado sintetizar una serie de derivados de triazina y bencimidazol que han mostrado actividad inhibidora del crecimiento de células cancerígenas (figura 1.13): R. Banerjee, D. R. Brown, E. Weerapana, SynLet., 2013, 24, 1599-1605. S. Kumar, H. R. Bhat, M. K. Kumawat, U. P. Singh, New J. Chem., 2013, 37, 581-584 y referencias allí citadas. 21 E. A. Peterson, P. S. Andrews, X. Be, A. A. Boezio, T. L. Bush, A. C. Cheng, J. R. Coats, A. E. ColLet.i, K. W. Copeland, M. DuPont, R. Graceffa, B. Grubinska, J.-C. Harmange, J. L. Kim, E. L. Mullady, P. Olivieri, L. B. Schenkel, M. K. Stanton, Y. Teffera, D. A. Whittington, T. Cai, D. S. La, Bioorg. Med. Chem. Let.., 2011, 21, 20642070. 19 20 1. Introducción general 29 Figura 1.13. Derivado de 1,3,5-triazina anticancerígeno. Además del cáncer, también pueden ser tratadas otras enfermedades con triazinas que presentan actividad citotóxica. De entre estas enfermedades cabe destacar la malaria, que actualmente es una de las enfermedades parasitarias más devastadoras en los países con menor desarrollo. Se estima que cada año se ven afectadas por esta enfermedad más de 300 millones de personas en África, Asia y Sudamérica, muriendo entre 1 y 3 millones. Los causantes de la enfermedad son protozoos parásitos del género Plasmodium, siendo el Plasmodium falciparum la especie causante de la mitad de todos los casos clínicos. Se han probado numerosos compuestos para luchar contra los parásitos de la malaria, algunos de los cuales han tenido éxito, como la cloroquina. El principal problema de estos compuestos es que los protozoos van desarrollando resistencia y se hacen necesarios tratamientos alternativos. En esta línea, se han sintetizado 4-aminoquinolintriazinas,22 como la que se muestra en la figura 1.14, que han mostrado actividad antimalárica. Figura 1.14. Derivado de 1,3,5-triazina antimalárico. Otro uso de los derivados de 1,3,5-triazina es como filtros UV en productos antisolares, que se incluye en este apartado porque en Estados Unidos los ingredientes 22 a) S. Manohar, S. I. Khan, D. S. Rawat, Bioorg. Med. Chem. Let.., 2010, 322. b) H. R. Bhat, U. P. Singh, P. Gahtori, S. K. Ghosh, K. Gogoi, A. Prakash, R. K. Singhm, New J. Chem., 2013, 37, 2654-2662. 30 1.3. Importancia de las 1,3,5-triazinas activos de estas cremas se consideran como fármacos.23 Se sabe que la radiación UV es responsable de una amplia variedad de efectos agudos y crónicos en la piel, tanto positivos (como la síntesis de vitamina D) como negativos, como eritemas (efecto agudo) o cáncer de piel (efecto a largo plazo). Existen patentes24 en las que se describe la actividad como protector solar de compuestos basados en el anillo de 1,3,5-triazina, con la estructura general que se muestra en la figura 1.15. Figura 1.15. Estructura general del principio activo en cremas solares. 1.3.2. HERBICIDAS Las triazinas fueron introducidas como herbicidas en 1954.25 El primer producto ensayado, la clorazina, se utilizó con éxito en la destrucción de la vegetación espontánea que crece en cultivos de algodón, tomate, maíz, cebollas, patatas o zanahorias. Posteriormente, se han introducido otras triazinas con marcado carácter herbicida; entre ellas conviene señalar la Simazina, ya que su uso estaba muy extendido en los olivares españoles,26 la Atrazina y la Propazina, mostrándose las estructuras de estos y otros herbicidas basados en el anillo de triazina en la figura 1.16. La actividad más importante de las triazinas es la destrucción de plantas en los primeros estados de desarrollo, de diez a quince días después de la germinación de las semillas, interfiriendo en el proceso de absorción de CO2 y formación de almidón. Estos herbicidas destruyen una amplia gama de hierbas tanto anuales como perennes; entre ellas podemos destacar el abrojo grande, la ortiga, la verdolaga, la amapola o el trébol. También se utilizan para el control selectivo de algas y malas hierbas submarinas en estanques, acuarios, fuentes y torres de recirculación de agua. 23 S. Nikolić, C. M. Keck, C. Anselmi, R. H. Müller, Int. J. Pharm., 2011, 414, 276-284. http://www.espatentes.com/pdf/2188883_t3.pdf 25 M. J. Higuera Camacho, Tesis Doctoral, 2003, pp 7-19. Universidad de Córdoba. 26 A. I. Cañero, L. Cox, S. Redondo-Gómez, E. Mateos-Naranjo, M. C. Hermosín, J. Cornejo, J. Agric. Food Chem., 2011, 59, 5528-5534. 24 1. Introducción general 31 Herbicida X R1 R2 Atrazina Cl -CH2-CH3 -CH-(CH3)2 Cianazina Cl -CH2-CH3 -C(CH3)2-CN Propazina Cl -CH-(CH3)2 -CH-(CH3)2 Cl -CH2-CH3 Simazina -CH2-CH3 Figura 1.16. Estructuras de herbicidas basados en el anillo de 1,3,5-triazina. A pesar de la efectividad de estos herbicidas, la eliminación de éstos del medio ambiente constituye un problema. Por ello, existe una cantidad ingente de trabajos de investigación cuyo objetivo es la eliminación de los residuos de herbicidas basados en el anillo de 1,3,5-triazina, recogiéndose algunos de ellos en una revisión de Krutz y colaboradores.27 1.3.3. QUÍMICA SUPRAMOLECULAR Las interacciones no covalentes juegan un papel importante en la determinación de la estructura y las propiedades de los agregados moleculares, que son primordiales en química, biología y ciencia de los materiales. Por este motivo, Mooibroek y Gamez28 nos hablan de la importancia del anillo de 1,3,5-triazina como tectón en química supramolecular. Destacan que este anillo puede intervenir en todas las interacciones intermoleculares descubiertas hasta la fecha, como puede verse en la figura 1.17, mencionando posteriormente algunas de ellas. L. J. Krutz, D. L. Shaner, M. A. Weaver, R. M. T. Webb, R. M. Zablotowicz, K. N. Reddy, Y. Huang, S. J. Thomson, Pest. Manag. Sci., 2010, 66, 461-481. 28 T. J. Mooibroek, P. Gamez, Inorg. Chim. Acta, 360, 2007, 381. 27 32 1.3. Importancia de las 1,3,5-triazinas Figura 1.17. Resumen de las interacciones supramoleculares en las que puede intervenir el anillo de 1,3,5-triazina. 1.3.3.1. Coordinación a metales: Los átomos de nitrógeno del anillo de triazina son nitrógenos de tipo piridínico, por lo que tienen un par de electrones libre, con el que son susceptibles de coordinar con metales. Therrien29 describe numerosos ejemplos de complejos basados en piridiltriazinas (figura 1.18). Figura 1.18. Isómeros simétricos de derivados de 2,4,6-tri(piridil)-1,3,5-triazina. A pesar de las similitudes entre los derivados de 2,4,6-tri(piridil)-1,3,5-triazina, la posición del nitrógeno de la piridina es importante para el modo de coordinación de los metales. Así, en el caso de 2,4,6-tri(pirid-2-il)-1,3,5-triazina (figura 1.18 a), tenemos la posibilidad de obtener complejos mono, di o trinucleares, dependiendo de la forma en 29 B. Therrien, J. Organomet. Chem., 2011, 696, 637-651. 1. Introducción general 33 la que este ligando se coordine con los átomos metálicos. Es interesante reseñar que, tras la coordinación del primer átomo metálico, pueden empezar a aparecer interacciones estéricas que condicionen la entrada de los siguientes átomos metálicos (figura 1.19). Figura 1.19. Impedimentos estéricos a la entrada de un segundo átomo metálico tras la coordinación del primero. En el caso de los isómeros b y c observamos diferentes estructuras, debido a que no existe la posibilidad de tener un ligando coordinado por tres puntos al átomo metálico. Algunos ejemplos de este tipo de coordinación se muestran en la figura 1.20. Figura 1.20. Ejemplos de complejos de derivados de 2,4,6-tri(piridil)-1,3,5-triazina con Cu (izquierda) y Mn (derecha). Por otro lado, Beller y colaboradores30 han descrito un complejo de 1,3,5triazina con iridio (figura 1.21) que puede funcionar como fotosensibilizador en la reacción de reducción del agua a hidrógeno catalizada por luz solar. 30 F. Gärtner, D. Cozzula, S. Losse, A. Boddien, G. Anilkumar, H. Junge, T. Schulz, N. Marquet, A. Spannenberg, S: Gladiali, M. Beller, Chem. Eur. J., 2011, 17, 6998-7006. 34 1.3. Importancia de las 1,3,5-triazinas Figura 1.21. Complejo de iridio que funciona como fotosensibilizador. 1.3.3.2. Enlaces de hidrógeno: La capacidad de los derivados de 1,3,5-triazina para formar supramoléculas mediante enlaces de hidrógeno está ampliamente descrita en la bibliografía,31 así como su capacidad para formar estructuras de tipo roseta (figura 1.22).32 Figura 1.22. Estructura de tipo roseta. La formación de un triple enlace de hidrógeno entre timina y melamina (1,3,5triaminotriazina), permitió a Huang y colaboradores33 desarrollar un nuevo método de detección de melamina en productos lácteos, usando politimina estabilizada con nanopartículas de oro. Este procedimiento, altamente sensible y selectivo, permite detectar la adulteración con melamina de los productos lácteos, evitando intoxicaciones a) P. Gamez, J. Reedijk, Eur. J. Inorg. Chem., 2006, 29; b) R. Wang, C. Pellerin, O. Lebel, J. Mater. Chem., 2009, 19, 2747; c) F. Vera, J. Barberá, P. Romero, J. L. Serrano, M. B. Ros, T. Sierra, Angew. Chem. Int. Ed., 2010, 49, 4910; d) J. Barber, L. Puig, P. Romero, J. L. Serrano, T. Sierra, J. Am. Chem. Soc., 2005, 127 (1), 458; e) A. Delori, E. Suresh, V. R. Pedireddi, Chem. Eur. J., 2008, 14, 6967; f) T. Seki, S. Yagai, T. Karatsu, A. Kitamura, J. Org. Chem., 2008, 73 (9), 3328; g) S. Yagai, S. Kubota, K. Unoike, T. Karatsu, A. Kitamura, Chem. Commun., 2008, 4466. 32 G. M. Whitesides, J. P. Mathias, C. T. Seto, Science, 1991, 254, 1312. 33 W. J. Qi, D. Wu, J. Ling, C. Z. Huang, Chem. Commun., 2010, 46, 4893. 31 1. Introducción general 35 (figura 1.23). Por otro lado, es interesante destacar los cálculos realizados por Wuest y Rochefort,34 los cuales revelan que las aminotriazinas tienen una fuerte afinidad por el grafito y sugieren que parte de la fuerza conductora para la adsorción es una interacción atractiva específica entre los grupos NR2 con la superficie. Estas interacciones distorsionan la capacidad formadora de enlaces de hidrógeno de estos compuestos. Figura 1.23. Esquema del procedimiento de detección de melamina en derivados lácteos. 1.3.3.3. Apilamiento: El apilamiento, en el plano vertical, a través de “interacciones π-π” y la extensión en el plano horizontal a través de enlaces de hidrógeno son los dos tipos de interacciones que prevalecen en los ensamblajes moleculares. Estas interacciones juegan un papel importante en el empaquetamiento de cristales que contienen partes aromáticas, estabilizando las grandes estructuras helicoidales tridimensionales de ADN y ARN, en química supramolecular y en procesos de reconocimiento intermolecular, entre otros. A continuación se muestran algunos ejemplos donde puede apreciarse la existencia de estas interacciones. 34 J. D. Wuest, A. Rochefort, Chem. Commun., 2010, 46, 2923. 36 1.3. Importancia de las 1,3,5-triazinas Mishra y colaboradores35 han realizado un estudio computacional sobre este tipo de interacciones para los anillos de 1,3,5-triazina, mostrando en la figura 1.24 un resumen de sus experiencias. Figura 1.24. Interacciones de apilamiento en 1,3,5-triazina. Hisamatsu y Aihara36 sintetizaron pinzas moleculares con 2,4,6-trifenil-1,3,5triazinas como espaciadores que exhiben dimerización a través de interacciones π-π stacking en estado sólido. Estas especies diméricas forman redes supramoleculares altamente organizadas, como se aprecia en la figura 1.25. Figura 1.25. Estructura por rayos X del dímero (a) y de la disposición molecular (b). Kawamichi y colaboradores37 han logrado construir una red cristalina basada en B. K. Mishra, J. S. Arey, N. Sathyamurthy, J. Phys. Chem. A, 2010, 114, 9606-9616. Y. Hisamatsu, H. Aihara, Chem. Commun., 2010, 46, 4902. 37 T. Kawamichi, T. Haneda, M. Kawano, M. Fujita, Nature, 2009, 461, 633. 35 36 1. Introducción general 37 una triazina, una amina e iones de cinc, que actúan como nodos (figura 1.26). Los autores llevaron a cabo la reacción de formación de una base de Schiff dentro de la red cristalina a baja temperatura (esquema 1.1). Para ello, primero atraparon el reactivo con un grupo funcional amina (representado en color verde en la figura 1.26) como molécula huésped en la red cristalina. Seguidamente, estabilizaron y observaron la estructura cristalina de un intermedio hemiaminal obtenido al hacer pasar un aldehído a través del cristal. Figura 1.26. Red cristalina basada en 1,3,5-triazina. Esquema 1.1. Reacción de formación de la base de Schiff que tiene lugar en la red cristalina. 1.3.4. DISPOSITIVOS OPTOELECTRÓNICOS Los compuestos basados en el anillo de 1,3,5-triazina han encontrado aplicaciones en el campo de los dispositivos optoelectrónicos, como es el caso de los diodos orgánicos emisores de luz (OLEDs), las células solares o en óptica no lineal. A continuación explicaremos brevemente en qué consiste cada campo y cómo se han hecho un hueco los compuestos basados en el anillo de 1,3,5-triazina. 1.3.4.1. OLEDs: Los materiales transportadores de electrones que forman parte de los OLEDs contienen frecuentemente heterociclos electrodeficientes. Dentro de estos heterociclos 38 1.3. Importancia de las 1,3,5-triazinas electrodeficientes, se sabe que las triazinas son buenos conductores de electrones, por lo que se han usado como capas transportadoras de electrones en OLEDs.38,39 Strohriegl y colaboradores40 proporcionan las triazinas que se muestran en la figura 1.27 como parte de OLEDs fosforescentes en el azul. Figura 1.27. Componentes de OLEDs fosforescentes en el azul. Por otro lado, se ha publicado que la 2,4,6-tris[p-(di-2-piridilaminofenil)]-1,3,5triazina (ver figura 1.28) emite en el azul, tanto en disolución como en estado sólido.41 Esta característica se atribuye al hecho de que los grupos amino en posiciones 2, 4 y 6 son donadores, dando lugar a una eficiente transferencia electrónica en el compuesto. λ = 440 nm; ФF = 0,78 Figura 1.28. 2,4,6-tris[p-(di-2-piridilaminofenil)]-1,3,5-triazina. S. Ren, D. Zeng, H. Zhong, Y. Wang, S. Qian, Q. Fang, J. Phys. Chem. B, 2010, 114, 10374. A. Richard, H. A. Klenklera, A. Tranc, D. Z. Popovic, G. Xu, Org. Elect. 2008, 9, 285. 40 M. M. Rothmann, S. Haneder, E. Da Como, C. Lennartz, C. Schildknetch, P. Strohriegl, Chem. Mater., 2010, 22, 2403. 41 J. Pang, Y. Tao, S. Frieberg, X-P. Yang, M. D’iorio, S. Wang. J. Mater. Chem. 2002, 12, 206. 38 39 1. Introducción general 1.3.4.2. 39 Óptica no lineal: Los materiales orgánicos muestran respuestas de óptica no lineal grandes y son de gran interés en el desarrollo de tecnologías fotónicas y optoelectrónicas, debido a su tiempo de respuesta rápido, gran susceptibilidad no lineal y coste de fabricación relativamente bajo. Los compuestos basados en el anillo de 1,3,5-triazina poseen buenas propiedades ópticas y electrónicas debido a su alta afinidad electrónica y estructura simétrica. Particularmente, las moléculas octupolares consistentes en un centro de triazina fuertemente electroatractor y un grupo electrodonador, unido a través de un puente π-conjugado, como la estructura que se muestra en la figura 1.29, han demostrado tener buenas propiedades de absorción de dos fotones.42 Figura 1.29. Estructura que experimenta absorción de dos fotones. Y. Jiang, Y. Wang, B. Wang, J. Yang, N. He, S. Qian, J. Hua, Chem. Asian J., 2011, 6, 157-165 y referencias citadas en la nota 1 de este artículo. 42 40 1.3. Importancia de las 1,3,5-triazinas Las prometedoras aplicaciones de los materiales orgánicos con absorción de dos fotones (2PA) en limitadores ópticos, láseres, microfabricación, almacenamiento de datos ópticos tridimensionales, bioimagen y terapia fotodinámica han atraído una atención considerable durante los últimos años.42 Estas aplicaciones estimularon la investigación en relaciones estructura-propiedades. El reto es cómo sintetizar los materiales con una gran sección de cruce de absorción de dos fotones. Algunas estrategias de diseño molecular eficientes han propuesto directrices de cara al desarrollo de materiales con grandes secciones cruzadas de absorción de dos fotones, incluyendo moléculas de tipo dador-aceptor-dador (D-AD), dador-puente π-aceptor (D-π-A), dador-π-dador (D-π-D), macrociclos, dendrímeros, polímeros y moléculas muy ramificadas. Estos estudios revelan que, extendiendo el sistema conjugado molecular para transferir carga o incorporar cromóforos multidipolares o cuadrupolares en una estructura molecular, incrementará el valor de la sección de un compuesto mientras se mantiene la transparencia lineal sobre un amplio rango del espectro.43 Los derivados de 1,3,5-triazina han encontrado también aplicación como limitadores ópticos. Un limitador óptico es un dispositivo óptico que experimenta alta transmisión hasta una cierta intensidad de entrada, cambiando a baja transmisión a partir de ese límite. Este comportamiento ofrece sensores y protección de ojos de la radiación láser en un amplio rango de longitudes de onda, previsto el amplio rango de longitudes de onda láser existentes.44 1.3.4.3. Células solares: Las células solares sensibilizadas por un colorante (DSSCs)45 han sido investigadas durante las últimas décadas como la tercera generación de células solares, debido a su facilidad de fabricación y bajo coste de producción. De hecho, se ha 42 Y. Jiang, Y. Wang, B. Wang, J. Yang, N. He, S. Qian, J. Hua, Chem. Asian J., 2011, 6, 157-165 y referencias citadas en la nota 1 de este artículo. 43 J. Liu, K. Wang, X. Zhang, C. Li, X. You, Tetrahedron, 2013, 69, 190-200. 44 M. A. Özdağ, T. Ceyhan, H. G. Yaglioglu, A. Elmali, Ö. Bekaroğlu, Opt. & Laser Tech., 2011, 992-995 45 Dye Sensitized Solar Cells. 1. Introducción general 41 demostrado que los complejos de DSSCs con rutenio presentan altas eficiencias de conversión fotoeléctrica. Pero, como el rutenio es un metal raro y caro, estos complejos no son económicamente sostenibles ni competitivos. Por este hecho es importante investigar en colorantes libres de metales para aplicaciones prácticas en DSSCs. Recientemente, se ha informado de la existencia de colorantes dador-π-aceptor (D-π-A) como excelentes fotosensibilizadores para DSSCs, debido a su alto coeficiente de extinción molar, longitud de onda de absorción modificable, síntesis sencilla y bajo coste. Dentro de este tipo de compuestos, Liu y colaboradores46 han logrado sintetizar una serie de sensibilizadores basados en el anillo de 1,3,5-triazina, como el que se muestra en la figura 1.30. Figura 1.30. Ejemplo de fotosensibilizador. 1.3.5. CIENCIA DE LOS MATERIALES Los compuestos basados en el anillo de 1,3,5-triazina han encontrado aplicación en numerosas ramas de la ciencia de los materiales. De entre estas ramas, destacaremos algunas como cristales líquidos, catalizadores o nanotecnología. 1.3.5.1. Cristales líquidos: Los enlaces de hidrógeno y las interacciones π-π se emplean con frecuencia como fuerzas conductoras para dar arquitecturas supramoleculares bien definidas. La melamina y sus derivados (2,4,6-triarilamino-1,3,5-triazinas), que pueden estar involucrados en ambos tipos de interacciones, han provisto una variedad de aproximaciones elegantes para diseñar nuevos tipos de materiales suaves. Se ha visto J. Liu, K. Wang, F. Xu, Z. Tang, W. Zheng, J. Zhang, C. Li, T. Yu, X. You, Tetrahedron Let.., 2011, 52, 6492 y referencias citadas. 46 42 1.3. Importancia de las 1,3,5-triazinas que las mesofases columnares que forman estos compuestos poseen potenciales aplicaciones para dispositivos electrónicos, como materiales y sensores químicos semiconductores y fotoconductores, debido a su alta movilidad transportadora de carga a lo largo de ejes columnares. Mostramos en la figura 1.31 un ejemplo de estructura de 1,3,5-triazina que forma parte de un cristal líquido, descrito por Cheng y colaboradores.47 Figura 1.31. Estructura óptica de la mesofase columnar del derivado de 1,3,5-triazina mostrado. 1.3.5.2. Catalizadores: Se ha descrito que algunos derivados de 1,3,5-triazina pueden presentar actividad catalítica. Por ejemplo, moléculas como el cloruro de cianurilo48 o derivados de melamina49 pueden actuar como catalizadores. 1.3.5.3. Nanotecnología: Los compuestos derivados de 1,3,5-triazina han encontrado también aplicaciones interesantes en el campo de la nanotecnología. Así, por ejemplo, Usachov y colaboradores50 han logrado dopar grafeno con nitrógeno usando una molécula de 1,3,5triazina, previamente depositada sobre una superficie de Ni (111). Un esquema representativo del proceso lo podemos ver en la figura 1.32. a)X. Cheng, J. Jin, Q. Li, X. Dong, Chin. J. Chem., 2010, 28, 1957-1962. b) H. K. Dambal, C. V. Yelamaggad, Tetrahedron Let.., 2012, 53, 186-190. 48 M. Tatina, S. K. Yousuf, D. Mukherjee, Org. Biomol. Chem., 2012, 10, 5357-5360. 49 E. A. Prasetyanto, M. B. Ansari, B.-H. Min, S.-E. Park, Catalysis today, 2010, 252-257. 50 D. Usachov, O. Vilkov, A. Grüneis, D. Haberer, A. Fedorov, V. K. Adamchuk, A. B. Preobrajenski, P. Dudin, A. Barinov, M. Oehzelt, C. Laubschat, D. V. Vyalikh, Nano Let.., 2011, 11, 5401-5407. 47 1. Introducción general 43 Figura 1.32. Grafeno dopado con nitrógeno. Wuest y Rochefort34 muestran, mediante cálculos teóricos, que el grafeno tiene gran afinidad por las aminotriazinas, sugieriendo que la fuerza directriz de la adsorción se debe a una interacción atractiva específica entre la superficie y los grupos NR2. Aprovechando este conocimiento, Vázquez y colaboradores51 lograron un método eficaz para exfoliar grafito, obteniendo grafeno de pocas capas, tal como se muestra en la figura 1.33. Figura 1.33. Exfoliación de grafito con melamina. J. D. Wuest, A. Rochefort, Chem. Commun., 2010, 46, 2923-2925. V. León, M. Quintana, M. A. Herrero, J. L. G. Fierro, A. de la Hoz, M. Prato, E. Vázquez, Chem. Commun., 2011, 47, 10936-10938. 34 51 44 1.3. Importancia de las 1,3,5-triazinas 1.3.5.4. Otras aplicaciones: Además de las aplicaciones anteriormente descritas, los derivados de 1,3,5triazina han sido útiles en otros muchos campos, de los que mencionaremos los más importantes. Una de estas aplicaciones es como aditivos de lubricantes, donde Xiong y colaboradores52 han visto que los compuestos que se muestran en la figura 1.34 son capaces de reducir la fricción. Figura 1.34. Estructura general de los aditivos. Otra aplicación interesante consiste en la construcción de materiales de alta densidad energética, que podrían ser empleadas como explosivos o como fuente de energía alternativa, en el caso de que fuésemos capaces de liberar esta energía de forma controlada. Así, Yang y colaboradores53 realizan unos cálculos en los que exponen el gran potencial como material de alta densidad energética de la 2,4,6-trinitro-1,3,5triazina (figura 1.35), cuya síntesis supone un reto que la comunidad científica aún no ha sido capaz de superar. Figura 1.35. Estructura de 2,4,6-trinitro-1,3,5-triazina. 52 53 L. Xiong, Z. He, H. Xu, J. Lu, T. Ren, X. Fu, Lubr. Sci., 2011, 23, 33-40. K. Yang, Y. H. Park, S. G. Cho, H. W. Lee, C. K. Kim, H.-J. Koo, J. Comput. Chem., 2010, 31, 2483-2492. 1. Introducción general 45 También podemos encontrar derivados de 1,3,5-triazina que se han empleado en almacenamiento de gases, principalmente formando parte de macroestructuras covalentes, como la que se muestra en la figura 1.36.54 Figura 1.36. Polímero empleado en almacenamiento de gases. 54 H. Lim, M. C. Cha, J. Y. Chang, Macromol. Chem. Phys., 2012, 213, 1385-1390. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 49 2. SÍNTESIS DE MONO Y BISTRIAZINAS. ESTUDIO DE SUS PROPIEDADES ÓPTICAS. 2.1. OBJETIVOS: Como primer objetivo, nos planteamos la síntesis de nuevos derivados de aminotriazinas π-conjugadas, que combinan la presencia de sistemas π-dadores y πaceptores (esquema 2.1). Los compuestos π-conjugados basados en el anillo de 1,3,5triazina se han estudiado como un candidato atractivo en materiales fotoeléctricos funcionales, debido a las interesantes propiedades estructurales y electrónicas de fragmento de triazina. Para ello, se utilizarán condiciones de reacción medioambientalmente benignas, destacando el uso de la radiación microondas como método de calefacción y la realización de la reacción sin disolvente. Asimismo, se intentará simplificar el procedimiento de purificación. Esquema 2.1. Síntesis de sistemas D-A-D con espaciadores π. Un segundo objetivo es el estudio de las propiedades ópticas de los compuestos obtenidos para evaluar sus posibles aplicaciones en dispositivos optoelectrónicos. Otro objetivo es la ampliación del sistema conjugado, sintetizando bistriazinas con un espaciador π, buscando mejorar las propiedades ópticas de estos derivados. 50 2.2. Antecedentes bibliográficos 2.2. ANTECEDENTES BIBLIOGRÁFICOS Debido a la importancia de los compuestos basados en el anillo de 1,3,5-triazina, hay numerosos grupos de investigación especializados en la síntesis de este tipo de compuestos. En este trabajo se muestran algunos resultados recientes de síntesis de derivados de triazina bajo irradiación microondas. La síntesis de derivados de triazina se ha llevado a cabo, fundamentalmente, mediante dos estrategias sintéticas claramente diferenciadas: reacciones de ciclación y reacciones de sustitución nucleófila sobre el cloruro de cianurilo (figura 2.1). 2ª sustitución 25 ºC R3 Cl N Cl N 1ª sustitución 0ºC NUCLEÓFILO BASE N Cl Y N R1 X R2 R4 N N Z R6 R5 X, Y, Z= C, N, O, S Rn= alquil, aril, alquenil 3ª sustitución 65 ºC Figura 2.1. Sustitución nucleófila sobre el cloruro de cianurilo. En lo que respecta a las reacciones de ciclación, Yadav55 ha realizado reacciones de ciclocondensación a partir de bases de Schiff derivadas de tiazol y aldehídos aromáticos en ausencia de disolvente, formando tiazolo-s-triazinas, como podemos ver en el esquema 2.2. S S N Ar1 N + Ar 2 NH4OAc + 3 Ar CHO MO, 6-12 min 75-89 % N Ar N 1 Ar3 Ar2 NH H H Esquema 2.2. Síntesis de tiazolo-s-triazinas. Otra reacción de ciclación es la que Dandia y colaboradores56 llevaron a cabo entre formaldehido acuoso y anilinas sustituidas con haluros o grupos trifluorometilo a) L. D. S. Yadav, R. Kapoor, Tetrahedron, 2003, 44, 8951 b)L. D. S. Yadav, S. Yadav, V. K. Rai, Green Chem., 2006, 8, 455. c) L. D. S. Yadav, V. K. Rai, S. Yadav, Let.. Org. Chem., 2007, 4, 47. 56 A. Dandia, K. Arya, M. Sati, P. Sarawgi, J. Fluor. Chem., 2004, 125, 1273. 55 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 51 bajo irradiación microondas, como se muestra en el esquema 2.3. Las hexahidrotriazinas resultantes presentan propiedades fungicidas. Esquema 2.3. Hexahidrotriazinas con propiedades fungicidas. En nuestro grupo de investigación se ha descrito la síntesis de 2,4-diamino1,3,5-triazinas-6-sustituidas por reacción de alquil-, aril- y heteronitrilos con dicianodiamida en presencia de una base y 1 ml de DMSO. El empleo de la radiación microondas como método de calefacción nos permitió desarrollar un procedimiento rentable, sencillo, limpio y cuidadoso con el medio ambiente57 (esquemas 2.4 y 2.5). De hecho, se obtuvo la bistriazina del esquema 2.5 con un 80 % de rendimiento en sólo 10 minutos. Estos derivados han sido utilizados por Manzano y colaboradores58,59 como tectones en química supramolecular. Esquema 2.4. Reacción de nitrilos con dicianodiamida. A. Diaz-Ortíz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, M. Mateo, A. Sánchez-Migallón, G. Valiente, New. J. Chem., 2004, 28, 952. 58 B. R. Manzano, F. A. Jalón, M. L. Soriano, M. C. Carrión, M. P. Carranza, K. Mereiter, A. M. Rodríguez, A. de la Hoz, A. Sánchez-Migallón, Inorg. Chem. 2008, 47, 8957. 59 B. R. Manzano, F. A. Jalón, M. L. Soriano, A. M. Rodríguez, A. de la Hoz, A. Sánchez-Migallón, Cryst. Growth Des., 2008, 8, 5, 1585. 57 52 2.2. Antecedentes bibliográficos NH 2 CN + H N 2 N H N N NH CN CN KOH / DMSO N MO / 10 min N NH2 NH 2 N N NH 2 80 % Esquema 2.5. Reacción de o-dicianobenceno con dicianodiamida. Años después, este método sería utilizado por Shie y Fang60 para sintetizar sistemas similares. Otra reacción parecida a la anterior ha sido publicada por Chen y colaboradores.61 Partiendo de nuevo de dicianodiamida, realizan una reacción de ciclación con un éster, obteniendo un derivado de triazina. Por otro lado, en nuestro grupo se han sintetizado 1,3,5-triazinas62 simétricas mediante ciclotrimerización de nitrilos en ausencia de disolvente y bajo irradiación microondas en un corto periodo de tiempo, comparado con los largos tiempos de reacción que requiere la calefacción convencional -más de 24 horas- (esquema 2.6). O O N MO / 1 h O N CN Y(OTf) 3 N N N N N O Esquema 2.6. Ciclotrimerización de N-cianomorfolina. En cuanto a la sustitución nucleófila sobre cloruro de cianurilo, Arya y Dandia63 prepararon mediante esta estrategia compuestos basados en el anillo de triazina, como se muestra en el esquema 2.7, empleando una zeolita como catalizador ácido. J-J. Shie, J-M. Fang, J. Org. Chem. 2007, 72, 3141. H. Chen, P. Dao, A. Laporte, C. Garbay, Tetrahedron, 2010, 51, 3174. 62 A. Díaz-Ortiz, A. de la Hoz, A. Moreno, A. Sánchez-Migallón, G. Valiente, Green Chem., 2002, 4, 339. 63 K. Arya, A. Dandia, Bioorg. Med. Chem. Let. 2007, 17, 3298. 60 61 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 53 Esquema 2.7. Trisustitución de cloruro de cianurilo. En este mismo sentido, en nuestro laboratorio se han sintetizado triazinas trisustituidas64 por reacción de aminas con cloruro de cianurilo usando la radiación microondas en ausencia de disolvente (esquema 2.8). Debemos recordar que la preparación de las triazinas trisustituidas en condiciones clásicas requiere condiciones fuertes de reacción, es decir, altas temperaturas y tiempos largos de reacción. Pero si se usa la radiación microondas, la reacción se lleva a cabo en sólo 10 minutos. Esquema 2.8. Trisustitución de cloruro de cianurilo. Además, se ha preparado una triazina soportada sobre un polímero, lo que amplía su aplicación en química combinatoria (esquema 2.9). NH Cl N HN N NH MO / 90 w / 10 min + HN NH 2 N N N N N N NH 135ºC / DMSO N N N N N N Esquema 2.9. Síntesis de una triazina soportada. A. Díaz-Ortiz, J. Elguero, A. de la Hoz, A. Jiménez, A. Moreno, S. Moreno, A. Sánchez-Migallón, QSAR Comb. Sci., 2005, 24, 649. 64 54 2.2. Antecedentes bibliográficos Otra reacción similar de sustitución nucleófila la realizan Kurteva y colaboradores65 absorbiendo los reactivos en gel de sílice, como se ve en el esquema 2.10. Esquema 2.10. Sustitución nucleófila sobre una monoclorotriazina. La reacción de 6-cloro-N,N’-bispirazolil-[1,3,5]-triazina-2,4-diaminas66 con 4aminobencilamina bajo irradiación microondas produce bistriazinas en excelentes rendimientos. El uso de una diamina que contiene grupos amino que presentan diferentes reactividades permitió llevar a cabo la reacción en dos pasos y dar selectivamente monotriazinas o bistriazinas que podían tener los sustituyentes iguales o diferentes (esquema 2.11). Estas bistriazinas nuevas tienen aplicaciones prometedoras en química supramolecular basada en enlaces de hidrógeno y/o complejación con metales. La presencia de una unión rígida puede usarse para una preparación eficiente de polímeros supramoleculares extendidos con propiedades fluorescentes interesantes mediante complejación con derivados de ácido cianúrico y ácido barbitúrico.67 Siguiendo con esta estrategia, irradiación con microondas, y usando como puentes derivados de diaminobenceno, se han obtenido una serie de derivados monoméricos de triazina con excelentes rendimientos. Asimismo, el método nos ha permitido sintetizar un conjunto de dímeros de 1,3,5-triazina como sistemas A-D-A πconjugados (esquema 2.12) que presentan interesantes propiedades fluorescentes.68 K. Doktorov, V. B. Kurteva, D. Ivanova, I. Timtcheva, ARKIVOC, 2007, XV, 232. A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente, Org. Biomol. Chem., 2003, 1, 4451. 67 M. Moral, A. Ruiz Carretero, M. I. López Solera, A. Sánchez-Migallón, A. de la Hoz, Tetrahedron, 2010, 66, 121127. 68 A. Ruiz Carretero, J. R. Ramírez, A. Sánchez-Migallón, A. de la Hoz, Eur. J. Org. Chem, submitted manuscript. 65 66 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 55 Esquema 2.11. Síntesis de mono y bistriazinas con puente 4-aminobencilamina. Esquema 2.12. Síntesis de mono y bistriazinas con fenilendiamina como espaciador. También se han sintetizado, mediante reacción de isocianato de fenilo con 2,4diamino-1,3,5-triazinas un conjunto de 2-amino-4-ureido-1,3,5-triazinas, que se podría dimerizar en una disposición autocomplementaria de cuatro enlaces de hidrógeno (figura 2.2), comprendidos de dos dadores (DD) y dos aceptores (AA). H O Ph H N N H N N H N N azol azol N N H N N H N N H H O Ph Figura 2.2. Interacciones supramoleculares en ureidotriazinas. 56 2.2. Antecedentes bibliográficos El comportamiento químico de los grupos amino unidos directamente al anillo de 1,3,5-triazina se parece más al de las amidas que al de las aminas. La radiación microondas en ausencia de disolvente permitió lograr la reacción de estas aminas, muy poco nucleófilas, con isocianato de fenilo, para formar selectivamente mono y bis ureidotriazinas (esquema 2.13).68 Esquema 2.13. Síntesis de mono y bisureidotriazinas. 68 A. Ruiz Carretero, J. R. Ramírez, A. Sánchez-Migallón, A. de la Hoz, Eur. J. Org. Chem, submitted manuscript. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 57 2.3. RESULTADOS OBTENIDOS Y DISCUSIÓN 2.3.1. TRIAZINAS CON 2,5-DIMETOXIANILINA: 2.3.1.1. Síntesis: Integrándome en la línea de nuestro grupo de investigación, comencé modificando los compuestos anteriormente descritos.67,68 Más concretamente, usamos como nucleófilo una anilina con dos grupos dadores metoxilo para reforzar el sistema dador y modificar las propiedades ópticas de los compuestos (esquema 2.14). Esquema 2.14. Esquema general de síntesis. La obtención de estructuras con disposición D-π-A-D es posible gracias a la unión del anillo de triazina (π-aceptor) a sustituyentes dadores, a través de espaciadores π-conjugados. Es interesante destacar que el compuesto 3a contiene dos anillos de pirazol. Este heterociclo es muy interesante, ya que aparte de ser un heterociclo π-excedente y, por tanto, electrodonador, tiene un nitrógeno en posición 2 que posee un par de electrones libre, que puede coordinarse a metales o tomar parte en enlaces de hidrógeno. M. Moral, A. Ruiz Carretero, M. I. López Solera, A. Sánchez-Migallón, A. de la Hoz, Tetrahedron, 2010, 66, 121127. 68 A. Ruiz Carretero, J. R. Ramírez, A. Sánchez-Migallón, A. de la Hoz, Eur. J. Org. Chem, submitted manuscript. 67 58 2.3. Discusión de resultados Además, se ha llevado a cabo la síntesis de derivados con grupos fuertemente electrodonadores (metoxilo) y heterociclos alifáticos, como piperidina y morfolina, que se unen al anillo de triazina mediante un átomo de nitrógeno que posee un par de electrones libres. Esto permitirá hacer una comparación de las propiedades de los diversos derivados obtenidos, con distintos grupos dadores. Para poner a punto el método experimental, se eligió el derivado de opirazolilfenilo (1a), previamente sintetizado en el grupo de investigación (esquema 2.15).66 Esquema 2.15. Obtención de 1a. Algunos de los resultados obtenidos para la síntesis del derivado 3a se encuentran recogidos en la tabla 2.1. A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente. Org. Biomol. Chem. 2003, 1, 4451. 66 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 59 Tabla 2.1. Síntesis de 3a. Cl N N N H O N N NH2 N M.O. O N H N N H3C N O 1a 2 Entrada a NH O N CH3 N H N N N N H N N 3a Tiempo (min) Temperatura (ºC) Disolvente Rto (%) 1 5 150 DMSO - 2 5 150 No 80 3 10 150 No 85 4 15 150 No 90 a) Relación 1a:2 = 1: 2 Todas las pruebas realizadas empleando DMSO dan lugar a mezclas difíciles de tratar, siendo imposible aislar el producto con un rendimiento aceptable. A modo de ejemplo se muestran los datos de una de las experiencias con disolvente y su comparación en las mismas condiciones sin DMSO (entradas 1 vs 2, tabla 2.1). En ausencia de disolvente se consiguieron los mejores resultados, alcanzando el 90 % de rendimiento en sólo 15 minutos (entrada 4). La principal dificultad se encontró a la hora de aislar y purificar el producto de reacción, para lo cual se realizaron diferentes intentos. Trabajar con derivados de triazina no es sencillo ya que, debido a la existencia de un elevado número de heteroátomos, las numerosas interacciones con la fase estacionaria hacen que la purificación por columna cromatográfica no siempre sea viable. Este hecho deja como principal recurso la purificación por solubilidad. En una de las pruebas se adicionó diclorometano (3 ml) al matraz de reacción, y se observó la aparición de un sólido, lo que hizo suponer que se había limpiado el producto. Filtrado el sólido, resultó ser la 2,5-dimetoxianilina de partida protonada, de acuerdo al espectro de 1H-RMN (figura 2.3). 60 2.3. Discusión de resultados La comparación del espectro de 1H-RMN con el del compuesto 2 sin protonar, (tabla 2.2) muestra que, como era de esperar, la protonación produce un desapantallamiento de las señales, a excepción de la del grupo NH, que se intercambia con el agua del disolvente. Tabla 2.2. Compuesto 2, efecto de la protonación en 1H-RMN. δ(ppm) Compuesto NH OCH3 (2) OCH3 (5) H3 H4 H6 2 4’72 (fina) 3’68 3’60 6’66 6’04 6’24 2-H+ 3’46 (ancha) 3’81 3’70 7’09 6’86 6’99 Figura 2.3. Ampliación del espectro de 1H-RMN (DMSO-d6) de 2 protonado. Por tanto, se estudió la fase filtrada, viendo por 1H-RMN que en ésta aparece el producto de reacción, junto con algunas trazas del compuesto 2, tanto en la forma neutra como en la protonada; por ello, se realizó un percolado en una columna de filtración con gel de sílice (10 mm de diámetro y 12 mm de altura) usando acetato de etilo como eluyente, de esta manera el compuesto 2 protonado queda retenido. De nuevo, el espectro de 1H-RMN revela que el producto de la reacción está impurificado por trazas de la amina de partida 2 (figura 2.4). Para eliminar esta impureza (señalada en el espectro con color rojo), se lavó el producto con disoluciones a diferentes concentraciones de HCl, viendo después de cierta experimentación que la concentración óptima fue 1M. Para neutralizar el producto, se añaden 5 ml de una 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 61 disolución saturada de carbonato sódico. Este procedimiento permitió obtener el producto puro con los rendimientos que se muestran en la tabla 2.3. 2-H+ Figura 2.4. 1H-RMN del crudo de reacción. Una vez puesto a punto el método experimental, se llevó a cabo la síntesis del resto de los derivados. En todos los casos se alcanzaron rendimientos excelentes en tan solo 15 minutos y en ausencia de disolvente (tabla 2.3). Cabe destacar que, con los derivados de piperidina (3d) y morfolina (3e), se eliminaron las trazas de producto 2 en condiciones más suaves ya que la concentración de HCl necesaria fue 0,1 M. Tabla 2.3. Rendimientos de 2,5-dimetoxiaminotriazinas. R 2.3.1.2. 3 a b c d e Rto (%) 90 95 90 85 80 Determinación estructural: Todos los compuestos fueron caracterizados mediante resonancia magnética nuclear [RMN (1H, 13 C, gHSQC, COSY)], espectroscopia de infrarrojo, puntos de fusión y espectrometría de masas. 62 2.3. Discusión de resultados La asociación inter e intramolecular de los compuestos sintetizados hace que la rotación de los enlaces Ctriazina-N sea más lenta que la escala de tiempo de la RMN. Por este motivo, los espectros registrados a 25ºC muestran señales anchas. A temperatura elevada, se consigue la energía suficiente para que el proceso de rotación de los enlaces sea rápido en la escala de tiempo de la RMN, lo que nos permite observar las señales características de estos compuestos. En las figuras 2.5 y 2.6 se muestran ampliaciones de estos espectros. Los espectros de los diferentes productos se encuentran en el anexo 2 de esta memoria. Los espectros de 13 C presentan señales alrededor de 165 ppm, típicas del anillo de 1,3,5-triazina. Entre 100 y 160 ppm presentan señales de carbonos correspondientes a anillos aromáticos, mientras que las señales correspondientes a carbonos alifáticos, tanto de los grupos metoxi como de los heterociclos alifáticos, aparecen por debajo de Figura 2.5. Espectro de 1H-RMN de 3a a 25 ºC (DMSO). 1.09 0.67 0.59 1.23 1.23 1.30 0.40 1.10 0.57 2.34 1.00 60 ppm. 63 1.10 0.57 0.59 1.25 1.28 1.22 0.52 1.52 1.09 1.11 1.00 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. Figura 2.6. Espectro de 1H-RMN de 3a a 80 ºC (DMSO). En todos los espectros de protón se observan señales en la región de 9 ppm, que corresponden a grupos aminos secundarios unidos a la triazina, así como las señales de los grupos OCH3 entre 3’6 y 3’9 ppm. En los espectros de infrarrojo se observa la presencia de bandas correspondientes a la vibración de tensión de enlaces N-H en la zona próxima a 3400 cm-1. También se distinguen las bandas de vibración de tensión simétrica y asimétrica del grupo OCH3 entre 1000 y 1300 cm-1. En la zona que corresponde a la vibración de tensión de los enlaces C=C y C=N (entre 1450 y 1600 cm-1) se aprecian bandas intensas difíciles de asignar. En los estudios realizados para determinar el punto de fusión de los diferentes productos, se pudo observar que 3a, 3b y 3c, con sustituyentes aromáticos, descomponen aproximadamente a 180 ºC mientras que, para 3d y 3e, con heterociclos alifáticos, la descomposición se produce alrededor de 200 ºC. En los espectros de masas se observan los picos correspondientes con las masas moleculares de los productos, aunque en el espectro del compuesto 3a se ha observado 64 2.3. Discusión de resultados la presencia de un pico correspondiente a 2M + H+, lo que podría ser indicativo de que el compuesto tiende a agregarse. 2.3.1.3. Estudio de las propiedades ópticas: 2.3.1.3.1. Espectroscopia UV-visible: El estudio de las propiedades ópticas de los derivados de triazina sintetizados se llevó a cabo mediante espectroscopia UV-visible y fluorescencia a temperatura ambiente, siguiendo el protocolo descrito en la parte experimental para las medidas en disolución de diclorometano. Los espectros de absorción se representan en la figura 2.7. 3a UV 3b UV 3c UV 3d UV 3e UV 0,8 Absorbance 0,6 0,4 0,2 0,0 300 400 Wavelength (nm) Figura 2.7. Espectros UV de los diferentes productos (CH2Cl2, 10-5 M). Los resultados obtenidos se recogen en la tabla 2.4, y muestran que los máximos de absorción se encuentran en la región del UV, alrededor de 300 nm. Este hecho, junto con la alta absortividad molar se atribuye a transiciones π-π*.69 También se puede observar un desplazamiento al rojo en los espectros de los compuestos que contienen anillos aromáticos (3a, 3b y 3c), respecto de los que contienen anillos alifáticos (3d y 3e), resultados que están de acuerdo con la existencia de una mayor conjugación.40 La formación de un puente de hidrógeno intramolecular en el caso del derivado de o-pirazolilfenilo (3a) podría contribuir a aumentar la planaridad E. Beltrán, J. L. Serrano, T. Sierra, R. Giménez, Org. Let.. 2010, 12, 1404. M. M. Rothmann, S. Haneder, E. Da Como, C. Lennartz, C. Schildknetch, P. Strohriegl, Chem. Mater. 2010, 22, 2403. 69 40 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 65 de la molécula, aumentando con ello la conjugación y teniendo como última consecuencia un desplazamiento batocrómico mayor. Tabla 2.4. Máximos de absorción de los compuestos 3a-e. Compuestoa λabs (nm) λfluoresc (nm) Desplazamiento ΦF -1 [log ε] de Stokes (cm ) 277 [4’90] 340, 398 6602 0’0011 3a 271 [4’16] 343 7745 0’0007 3b 272 [4’43] 330 6461 0’005 3c 259 [4’544], 305 [4’27] 340, 405 3375 0’003 3d 259 [4’590], 305 [4’37] 340 3375 0’0012 3e a) Disoluciones 10-5 M en CH2Cl2. 2.3.1.3.2. Fluorescencia: Se denomina fluorescencia a la emisión de un fotón durante el proceso de relajación entre el estado excitado (S1) y el estado fundamental (S0) de la molécula. Puesto que la emisión siempre se da desde el estado S1, las características de la misma serán independientes de la longitud de onda de excitación. Este espectro de emisión se situará siempre a longitudes de onda mayores que el espectro de absorción, según la regla de Stokes. Este hecho se refleja en el espectro de fluorescencia, que se muestra en la figura 2.8, realizado al compuesto 3a. El desplazamiento de Stokes70 representa la pérdida de energía que una molécula sufre desde que es excitada hasta que relaja emitiendo luz. Esta pérdida de energía se produce frecuentemente de forma térmica y se debe a relajaciones de estados vibracionales y rotacionales, pero en ocasiones un valor de desplazamiento de Stokes mayor de 5000 cm-1 indica procesos más complejos de pérdida de energía.71 Así pues, los valores de Stokes que se observan (tabla 2.4) ponen de manifiesto que sí existe conjugación en el sistema, pero no llegan a ser lo suficientemente grandes como para suponer la existencia de un sistema de separación de cargas. El desplazamiento de Stokes se define como la diferencia de energía entre la longitud de onda más intensa en el espectro de absorción y la longitud de onda más intensa en el espectro de emisión. 71 Se considera que valores de Stokes por debajo de 5000 cm-1 corresponden a una pérdida de energía típica de relajaciones rotacionales y vibracionales. 70 66 2.3. Discusión de resultados 3a UV 3a Fluorescence 0,8 3,0 2,5 0,6 Absorbance 1,5 0,4 1,0 Intensity (a.u.) 2,0 0,2 0,5 0,0 300 400 500 0,0 600 Wavelength (nm) Figura 2.8. Espectros de UV y fluorescencia del derivado de fenilpirazol 3a (CH2Cl2, 10-5 M). Se puede observar cómo los espectros de fluorescencia de los derivados con sustituyentes alifáticos, piperidina 3d (figura 2.9) y morfolina 3e (figura 2.10), obtenidos por irradiación en los diferentes máximos del espectro de absorción, son similares, es decir, se obtienen espectros análogos independientemente del máximo de absorción que se excite. Esto se debe a un mecanismo de conversión interna, donde se produce la relajación de un electrón no enlazante de un nivel n hasta el nivel fundamental π. Asimismo, los espectros de fluorescencia que se muestran en las figuras 2.9 y 2.10 presentan una banda o un hombro intenso alrededor de 400 nm que asociamos a la presencia de excímeros en nuestra disolución. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 67 3d UV 3d Fluorescence 259 nm 3d Fluorescence 305 nm 0,25 2 Intensity (a.u.) Absorbance 0,20 0,15 0,10 0,05 0,00 0 300 400 500 600 Wavelength (nm) Figura 2.9. Espectros UV y fluorescencia de 3d (CH2Cl2, 10-5 M). 3 3e UV 3e Fluorescence 259 nm 3e Fluorescence 305 nm 0,7 0,6 2 0,4 0,3 1 Intensity (a.u.) Absorbance 0,5 0,2 0,1 0,0 300 400 500 0 600 Wavelength (nm) Figura 2.10. Espectros UV y fluorescencia de 3e (CH2Cl2, 10-5 M). Un excímero es un dímero que se forma en el estado excitado (del inglés excited dimer). Se forma por colisión entre una molécula excitada y una molécula iónica no excitada: 1 M* + 1M → 1(MM)* La representación simbólica (MM)* intenta describir que la energía de excitación se encuentra deslocalizada sobre los dos monómeros. Una vez se relaja la molécula, el 68 2.3. Discusión de resultados excímero se disocia. La banda correspondiente a un excímero se localiza a longitudes de onda mayores que la correspondiente al monómero y además nunca exhibe bandas vibrónicas.72 Se realizó un experimento para determinar si nuestra percepción inicial sobre el excímero era correcta. Consiste en realizar espectros de fluorescencia de un compuesto a diferentes concentraciones, debiendo obtener que la banda del excímero es más intensa para las disoluciones más concentradas mientras que, en las disoluciones más diluidas, la banda del monómero debe ser más intensa. En nuestro caso, hemos realizado los experimentos con el derivado de morfolina (figura 2.11) y el de pmetoxifenilo (figura 2.12) usando concentraciones entre 10-3 y 10-5 M, como se muestra a continuación. Estas experiencias nos permiten apreciar de forma clara que realmente tenemos bandas correspondientes a excímeros ya que, para las disoluciones más diluidas tienen algo más de intensidad las bandas más próximas a 340 nm (zona de emisión del monómero excitado), mientras que en la región cercana a 400 nm (zona donde se registra la emisión del excímero), la banda de la disolución más concentrada es más intensa, lo que está de acuerdo con la existencia de asociaciones en el estado excitado. -3 3e 10 M -4 3e 10 M -5 3e 10 M 1,0 Intensity (a.u.) 0,8 0,6 0,4 0,2 0,0 300 400 500 Wavelength (nm) Figura 2.11. Espectros de fluorescencia a diferentes concentraciones de 3e en CH2Cl2. 72 B. Valeur, “Molecular fluorescence. Principles and Applications”, Wiley-VCH, Weinheim (Alemania), 2002 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 69 -3 3c 10 M -4 3c 10 M -5 3c 10 M 1,0 Intensity (a.u.) 0,8 0,6 0,4 0,2 0,0 300 400 500 Wavelength (nm) Figura 2.12. Espectros de fluorescencia a diferentes concentraciones de 3c en CH2Cl2. 2.3.1.3.3. Excitación: La variación en la intensidad de fluorescencia en función de la longitud de onda de excitación (λE) para una longitud de onda fija de emisión (λF) se conoce como espectro de excitación. Cuando se dan varias especies o una sola especie presente diferentes formas en el estado fundamental (como, por ejemplo, agregados), el espectro de absorción y el de excitación no serían superponibles. Debido a esto, la comparación de estos espectros es útil, ya que nos puede ofrecer información valiosa. Así pues, en la tabla 2.5 se recogen los datos de los espectros de excitación y de absorción de los compuestos 3a, 3d y 3e, en la que se muestran las longitudes de onda correspondientes a los máximos en cada espectro. Tabla 2.5. Espectros de excitación y de emisión de los compuestos 3a, 3d y 3e. R 3a 3d 3e λabsorción (nm) λexcitación (nm) Máximos excitación (nm) 277 340 283 259, 305 259, 305 398 283 340 270, 306 405 283, 339 340 271, 304 70 2.3. Discusión de resultados El espectro de excitación realizado al derivado de morfolina se muestra en la figura 2.13. 3e UV 3e Excitation 340 nm 0,7 2,0 0,6 Absorbance 0,4 1,0 0,3 0,2 Intensity (a.u.) 1,5 0,5 0,5 0,1 0,0 0,0 300 Wavelength (nm) Figura 2.13. Espectros de UV y excitación 3e (CH2Cl2, 10-5 M). Aunque parece que hay desplazamiento de 271 nm a 259 nm del máximo de la banda de excitación al de la banda de absorción, el hecho de que el máximo del espectro de absorción se encuentre en el azul hace que la absorción del disolvente interfiera en la medida, por lo que es muy difícil comprobar la diferencia entre el espectro de ultravioleta y el de excitación, la banda a 305 nm que coincide en ambos espectros parece indicar la ausencia de agregados. Esta coincidencia también se pone de manifiesto en los espectros de excitación de los compuestos 3a y 3d (figuras 2.14 y 2.15, respectivamente). Sin embargo, en ambos casos cabe destacar: - Por un lado, para el derivado de pirazol 3a, el espectro de excitación es diferente a 340 nm y a 398 nm. A 340 nm coincide con el espectro de absorción y, por tanto, indica procesos de absorción y emisión normales. En cambio, a 398 nm el espectro de excitación no coincide con el de absorción. Esto significa que esta banda (398 nm) es un excímero. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 71 2,5 3a UV 3a Excitation 340 nm 3a Excitation 398 nm 0,8 2,0 Intensity (a.u.) Absorbance 0,6 1,5 0,4 1,0 0,2 0,5 0,0 0,0 300 400 Wavelength (nm) Figura 2.14. Espectros UV y excitación del producto 3a (CH2Cl2, 10-5 M). - Por otro lado, para el derivado de piperidina 3d tenemos un espectro de excitación a 405 nm que no es superponible con el espectro de ultravioleta, lo cual nos indica de nuevo la presencia de agregados moleculares. 2,5 3d UV 3d Excitation 340 nm 3d Excitation 405 nm 0,25 2,0 1,5 0,15 1,0 0,10 Intensity (a.u.) Absorbance 0,20 0,5 0,05 0,00 0,0 400 300 Wavelength (nm) Figura 2.15. Espectros UV y excitación del producto 3d (CH2Cl2, 10-5 M). 2.3.1.3.4. Influencia del disolvente: El término “solvatocromismo” se usa para describir el cambio pronunciado en la posición (y a veces en la intensidad) de una banda de absorción en el espectro UVvisible al variar la polaridad del medio (disolvente). Un desplazamiento batocrómico de 72 2.3. Discusión de resultados la banda, corresponde a un solvatocromismo positivo. Un desplazamiento hipsocrómico de la banda corresponde a un solvatocromismo negativo.73 Por ello, otro estudio realizado consistía en ver la influencia del disolvente en las propiedades ópticas de nuestros productos. Se eligió el derivado de fenilpirazol, usando como disolventes de diversa polaridad hexano, diclorometano y metanol. Los resultados obtenidos se recogen en la tabla 2.6. Tabla 2.6. Influencia del disolvente para el compuesto 3a Compuesto Hexano Diclorometano Metanol 276 277 272 Máximo de absorción 3a 348 339 398 Máximo de fluorescencia 3a Se querían hacer también estos experimentos usando disolventes como DMSO o acetona, pero el “cut-off”74 del DMSO es de 268 nm, demasiado cercano a los máximos de absorción (en los casos de las estructuras con anillos alifáticos unidos al núcleo de triazina es incluso superior este “cut-off” al máximo de absorción), mientras que con la acetona este parámetro es de 330 nm, bastante por encima de nuestro máximo de absorción, por lo que en este caso no son disolventes adecuados para realizar los experimentos de absorción UV-visible.75 Los resultados obtenidos nos permiten comentar, en primer lugar, que si hubiese separación de cargas, se observaría un desplazamiento al rojo de las bandas conforme aumenta la polaridad del disolvente, y no es nuestro caso. En segundo lugar, el disolvente puede afectar a la agregación de las moléculas, tanto por su polaridad como por su participación en enlaces de hidrógeno, hecho que se reflejaría en variaciones en la banda de excímeros y en la banda de fluorescencia normal. Esto es lo que se observa en la figura 2.16. A. García, B. Insuasty, M. A. Herranz, R. Martínez-Álvarez, N. Martín, Org. Let.. 2009, 11, 5398. En física teórica, “cut-off” es el valor máximo o mínimo de energía, momento o longitud, que nos indica que los objetos cuyas propiedades físicas queden fuera de esos límites serán ignorados. El “cut-off” en ultravioleta es la máxima energía permitida o la longitud de onda más pequeña permitida. 75 A. P. H. J. Schenning, P. Jonkheijm, E. Peeters, E. W. Meijer, J. Am. Chem. Soc. 2001, 123, 409. 73 74 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. DCM Hex MeOH 1,0 Intensity(a.u.) 73 0,5 0,0 300 400 500 600 Wavelength (nm) Figura 2.16. Espectros de fluorescencia de 3a con diferentes disolventes. 2.3.1.3.5. Estudios de agregación: La capacidad de estos compuestos, inherente a su estructura, para interaccionar a través de enlaces secundarios, así como la presencia de excímeros, nos ha estimulado a llevar a cabo estudios de agregación. En primer lugar, se ha usado el método de encapsulación con Rojo Nilo76 para determinar la concentración de agregación crítica (CAC en adelante) de estos productos. Este compuesto, cuya estructura se muestra en la figura 2.17a, presenta una longitud de onda de fluorescencia que varía dependiendo del medio en el que se encuentre (figura 2.17b).77 Si nuestros productos agregan, dichos agregados provocarían la encapsulación de la molécula de Rojo Nilo, haciendo que la fluorescencia de la disolución varíe. Esta variación se debería exclusivamente a la disminución de la concentración de Rojo Nilo libre, ya que nuestro producto no emite fluorescencia a las longitudes de onda estudiadas. En este caso, no observamos ningún cambio en la fluorescencia al (a)Y. B. Lim, E. Lee, M. Lee, Angew. Chem. Int. Ed., 2007, 46, 9011-9014. (b)M. C. A. Stuart, J. C. van de Pas, J. Engberts, J. Phys. Org. Chem. 2005, 18, 929-934. 77 http://en.wikipedia.org/wiki/Nile_red 76 74 2.3. Discusión de resultados incrementar la concentración de nuestro producto, lo que nos hace pensar que el Rojo Nilo no es capaz de introducirse entre los agregados moleculares. a) b) Figura 2.17. a) Estructura de la molécula de Rojo Nilo (9-dietilamino-5H-benzo[a]fenoxazin-5ona). b) Rojo Nilo bajo luz visible y UV en diferentes disolventes. De izquierda a derecha: agua, metanol, etanol, acetonitrilo, dimetilformamida, acetona, acetato de etilo, diclorometano, hexano, metiltercbutiléter, ciclohexano, tolueno. Una segunda aproximación es la realización de estudios de DOSY,78 pero las concentraciones necesarias para ejecutar estas experiencias son superiores a la solubilidad de nuestros productos. La tercera aproximación es recurrir a experimentos de Dynamic Light Scattering (DLS) para estudiar la presencia de agregados. DLS es una técnica que permite medir la distribución del tamaño de las partículas presentes en una disolución, rindiendo también el radio hidrodinámico de las mismas. En el caso reflejado en la figura 2.18, el diámetro hidrodinámico de la población más abundante de partículas sería de 38 nm. Las gráficas que muestran la distribución del tamaño de las partículas en disolución de los derivados 3a-e se muestran en el anexo 2. En la tabla 2.7 se recogen los resultados de los experimentos de DLS realizados a estos derivados, donde observamos agregación de estas comprendidos entre 38 y 296 nm. 78 Diffusion Ordered Spectroscopy. moléculas, con diámetros hidrodinámicos 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 75 Figura 2.18. Distribución del diámetro hidrodinámico de las especies en una disolución 10-3 M en THF del derivado de piperidina 3d. Tabla 2.7. Resultados de los experimentos de DLS. ¿Forma Diámetro agregados? hidrodinámico THF Sí 115 nm 10-2 M CH2Cl2 Sí 296 nm 3c 10-3 M CH2Cl2 No -- 3d 10-3 M THF Sí 38 nm 3e 10-2 M CH2Cl2 Sí 171 nm Compuesto Concentración Disolvente 3a 10-3 M 3b Del conjunto de estas experiencias cabe destacar que, aun a alta dilución (10-5 M) se observan procesos de agregación, donde las moléculas tienden a estar próximas por interacciones intermoleculares, como enlaces de hidrógeno, lo que hace que, aun a concentraciones tan bajas, se observen excímeros, o sea, la molécula que absorbe tiene 76 2.3. Discusión de resultados otra muy cerca para asociarse con ella. Estas agregaciones también se ponen de manifiesto con el cambio de disolvente (como hemos visto en la sección 2.3.1.3.4) y con los experimentos de DLS. Para finalizar, estos compuestos presentan unos rendimientos cuánticos bajos (tabla 2.4) por lo que no se pueden utilizar para la construcción de OLEDs. Tampoco se pueden emplear para construir células solares porque presentan muy poca transferencia de carga. Por ello, reorientamos nuestros objetivos a ampliar el sistema π con la síntesis de bistriazinas, haciendo las reacciones con anilinas difuncionalizadas, como 1,5diaminonaftaleno (figura 2.19).79 Figura 2.19. 1,5-diaminonaftaleno. 2.3.2. DERIVADOS DE TRIAZINA CON 1,5DIAMINONAFTALENO 2.3.2.1. Síntesis: Tratando de mejorar los resultados obtenidos en la sección anterior, y continuando con los métodos de síntesis sostenible desarrollados en nuestro grupo de investigación, abordamos la obtención de nuevos sistemas de naftilaminotriazina por reacción de sustitución nucleófila sobre un derivado de monoclorotriazina, que contiene sustituyentes tanto aromáticos como alifáticos. Como modificación del trabajo anteriormente descrito,69 se ha usado un puente que ya presenta propiedades fluorescentes, como es el 1,5-diaminonaftaleno que además, al estar difuncionalizado, nos permitirá sintetizar mono (5) y bistriazinas (6), siendo estas últimas nuestro verdadero objetivo ya que, al tener una mayor conjugación, nos permitirán conseguir mejores propiedades. (Esquema 2.16). M. El-Sedik, N. Almonasy, M. Nepraš, S. Bureš, M. Dvořák, M. Michl, J. Čermak, R. Hrdina, Dyes Pigments, 2012, 92, 1126-1131. 69 A. Ruiz Carretero, J. R. Ramírez, A. Sánchez-Migallón, A. de la Hoz, Eur. J. Org. Chem, submitted manuscript. 79 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 77 Esquema 2.16. Síntesis de mono y bistriazinas con 1,5-diaminonaftaleno. En esta ocasión, podemos obtener estructuras con disposición D-A-D, gracias a la unión del anillo de triazina (π-aceptor) con sustituyentes dadores, a través de espaciadores π-conjugados o, en el caso de las bistriazinas, tendríamos estructuras con disposición D-A-D-π-D-A-D. Con la idea de poner a punto un método experimental de síntesis, partimos del derivado de piperidina (1d), sintetizado en el grupo de investigación66 mediante la siguiente reacción (esquema 2.17). N Cl + N N Cl EtiPr2 N Cl Cl H N THF [Ar] 1) 0 ºC, 2h 2) TA, 24h N N N N N 1d Esquema 2.17. Síntesis del reactivo 1d. En primer lugar, exploramos la posibilidad de obtener selectivamente el derivado 5d. Así, la tabla 2.8 recoge un resumen de las experiencias realizadas. A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente. Org. Biomol. Chem., 2003, 1, 4451. 66 78 2.3. Discusión de resultados Tabla 2.8. Síntesis de 5d. Entrada Proporción 1d : 4 0’5 : 0’5 1 0’5 : 0’5 2 0’5 : 0’5 3 0’5 : 0’5 4 0’5 : 1 5 0’5 : 1 6 0’5 : 1 7 0’5 : 1’5 8 a. b. Temperatura (ºC) 150 170 170 185 180 180 180 180 Tiempo (min) 15 15 45 45 15 15 20 25 Disolvente Base DMSO DMSO DMSO DMSO No No No No DIPEA DIPEA DIPEA DIPEA No Na2CO3 No No 5d Rto. (%) --a --a --a --a --a --b --a 62 %. Producto no aislado. Producto no aislado. Por CCF se ve mezcla de 5d y 6d. Como condiciones iniciales de reacción (entrada 1, tabla 2.8) fijamos una temperatura de 150 ºC y un tiempo de 15 minutos. Como ambos reactivos son sólidos, usamos DMSO como disolvente y diisopropiletilamina (DIPEA) como base. Se observa por cromatografía en capa fina (CCF en adelante) que prácticamente no hay reacción, por lo que en sucesivas experiencias, se aumenta el tiempo o la temperatura (entradas 2 a 4, tabla 2.8), sin lograr obtener resultados positivos. Viendo que las reacciones con DMSO no son sencillas, decidimos probar un método sin disolvente. Para ello, debemos tener en cuenta el punto de fusión de nuestros reactivos. El 1,5-diaminonaftaleno funde a 186-188 ºC80 por lo que, teniendo en cuenta que las mezclas siempre funden a una temperatura sensiblemente inferior a la del reactivo puro, se fija como temperatura inicial 180 ºC y un tiempo de 15 minutos (entrada 5, tabla 2.8), poniendo un exceso de 1,5-diaminonaftaleno para que cumpla la función de base, además de la de nucleófilo. La reacción sale bastante limpia, según una cromatografía en capa fina realizada, ya que sólo aparece el producto 5d, trazas del reactivo 1d y parte de reactivo 4, que se pone en exceso. 80 J. Rodriguez, J. Gonzalo, J. L. Tejedor, J. Org. Chem., 2002 , 67, 22, 7631-7640. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 79 La inclusión de una base adicional en la reacción, como carbonato sódico (entrada 6, tabla 2.8), condujo a mezclas de productos, probablemente debido a que es mucho más difícil controlar la temperatura de reacción, por lo que se descartó este método. Buscando que la reacción se complete, se incrementó el tiempo de reacción (entradas 7 y 8, tabla 2.8), logrando el objetivo en el último caso. Una vez logrado que la reacción se complete, la purificación del producto no es evidente. Después de múltiples pruebas con diferentes disolventes, al añadir acetona a la mezcla de reacción, aparece un sólido, que resulta ser parte del producto 4 protonado. Por tanto, se filtró para eliminar este sólido y se añadieron los equivalentes necesarios de HCl a la mezcla de reacción para provocar la precipitación del exceso de 4 puesto en la reacción. Una vez hecho esto, el siguiente paso consistió en añadir agua a la mezcla, consiguiendo precipitar selectivamente el producto 5d protonado. Lavando con NaOH se obtuvo 5d puro con un 62 % de rendimiento. En la entrada 6 de la tabla 2.8 se observa que un aumento de la temperatura daba una mezcla de 5d y 6d. Por ello, se llevó a cabo la reacción a mayor temperatura para obtener directamente el derivado de bistriazina 6d, según se muestra en los datos recogidos en la tabla 2.9. Después de la optimización de la reacción, se fijaron como mejores condiciones 200 ºC y ausencia de disolvente, hecho importante desde el punto de vista de la sostenibilidad. 80 2.3. Discusión de resultados Tabla 2.9. Síntesis de 6d. Entrada 1 2 3 4 5 a. Proporción Temperatura (ºC) Tiempo (min) Disolvente 6d Rto. (%) 1d : 4: KOH 1 : 0’5 : 1 200 15 No a 1 : 0’5 : 1 200 45 No a 1’25 : 0’5 : 1 200 45 No a 1’25 : 0’5 : 1 200 60 No 67 1’1 : 0’5 : 1 200 60 No 72 Por CCF se ve mezcla de 5d:6d. Para empezar esta serie de reacciones, se fijó una temperatura inicial de 200 ºC y un tiempo de 15 minutos sin disolvente (entrada 1, tabla 2.9), poniendo un equivalente de KOH. Como observamos que la reacción no termina, es necesario aumentar el tiempo o la temperatura de reacción. Ya que la temperatura es bastante alta, y en previsión de que puedan descomponer nuestros productos, se decide mantenerla en 200 ºC y aumentar el tiempo de reacción. De esta manera, la entrada 2 muestra que el tiempo se ha incrementado hasta los 45 minutos, viendo que tenemos mezcla de los productos mono (5d) y bis-sustituido (6d), lo que nos indica que es necesario aumentar la proporción del reactivo 1d (entradas 3 vs 5). Las condiciones óptimas de reacción se muestran en la entrada 5. En cuanto a la etapa de purificación del producto, se observó que la solubilidad de la bistriazina 6d en acetona es baja, mientras que tanto 1d como 5d sí son solubles, por lo que simplemente se añade acetona al crudo de reacción y se filtra. De esta manera, obtenemos el derivado de bistriazina con un 72 % de rendimiento. Al tratar de extender el método sintético a otros derivados de mono y bis triazinil aminonaftalenos (ver esquema 2.17), nos hemos encontrado serias dificultades en el método de purificación, y no hemos sido capaces de aislar los productos deseados. Por ello, cambiamos la estrategia sintética, con el objetivo de obtener dichas bistriazinas. En 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 81 esta ocasión, se parte del precursor que se indica en la figura 2.20, descrito por Wei y colaboradores:81 Figura 2.20. Precursor de las bistriazinas. Nuestra idea es realizar una sustitución nucleófila sobre los cuatro enlaces C-Cl que presenta la molécula anterior (esquema 2.18). Así, como nucleófilos elegimos aminas de diferentes características, como pueden ser la morfolina, la p-anisidina, la pnitroanilina o la difenilamina, entre otras. Con ello pretendemos obtener productos con características diferentes para ver cuál de ellos nos da mejores propiedades. Esquema 2.18. Síntesis de N,N-bis-(1,3,5-triazin-2-il)-1,5-diaminonaftalenos. Para poner a punto el método experimental, elegimos el derivado de p-anisidina, con el objetivo de sintetizar la bistriazina 6c, principalmente porque no hemos sido capaces de aislarlo por el otro método. Los datos de las experiencias realizadas se recogen en la tabla 2.10. 81 W. Wei, H-J. Wang, C-Q. Jiang, Spectrochimica Acta Part A, 2008, 70, 362-366. 82 2.3. Discusión de resultados Tabla 2.10. Síntesis de 6c. Entrada Proporción Temperatura (ºC) Tiempo (min) 6b Rto. (%) 7 : 8b 0’25 :2 140 10 50 1 0’25 : 2 140 20 65 2 Como experiencia inicial (entrada 1, tabla 2.10), se emplean unas condiciones de temperatura de 140 ºC y un tiempo de 10 minutos, sin disolvente (el punto de fusión de la p-anisidina, según el catálogo del proveedor, es de entre 56 y 59 ºC, con lo que la reacción transcurriría en fase fundida. Se utiliza una proporción de los reactivos de 1:8 porque la anilina 8c actuará de nucleófilo y de base, debido a que se desprende HCl en la reacción. Tras lavar con una disolución de HCl el crudo de reacción, para eliminar los restos de anilina de partida, observamos dos manchas en la CCF, lo que nos hace pensar que la reacción no ha acabado. Diferentes experiencias donde se va prolongando el tiempo de reacción nos permiten completar la misma en sólo 20 minutos (entrada 2, tabla 2.10). Previamente se había probado temperaturas de alrededor de 160 ºC, observándose por CCF mezclas de reacción mucho más complejas. Para purificar el producto, probamos a realizar lavados. En primer lugar, como ya se ha mencionado anteriormente, se añade una disolución de HCl a los crudos para eliminar el exceso de anilina que pueda quedarnos. Seguidamente, probamos a añadir diferentes disolventes a nuestro crudo, tratando de que nos disuelvan sólo las impurezas, quedándonos nuestro producto como precipitado. En este caso, los disolventes adecuados resultan ser acetona o diclorometano. Los espectros de RMN nos confirman que la estructura del sólido obtenido corresponde con la del producto esperado. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 83 A continuación, se extiende esta metodología para las aminas anteriormente mencionadas. En la tabla 2.11 se recogen las mejores condiciones para cada uno de los casos. Tabla 2.11. Resultados obtenidos para la síntesis de bistriazinas. R 6b 6c 6e 6f Temperatura Tiempo 6 (ºC) (min) Rto. (%) 140 10 85 140 20 65 120 10 85 200 10 40 150 10 60 6h Proporción: 7: 8b-h: 0’25 mmol: 2 mmol. 2.3.2.2. Determinación estructural: Todos los compuestos fueron caracterizados mediante resonancia magnética nuclear [RMN (1H, 13 C, gHSQC, COSY)], espectroscopia de infrarrojo, puntos de fusión y espectrometría de masas. 84 2.3. Discusión de resultados En todos los espectros de 1H se observan señales en la región de 9 ppm, que corresponden a grupos aminos secundarios unidos a la triazina. Por otro lado, para el caso de la monotriazina 5d, se puede apreciar la señal del grupo amino libre a un desplazamiento químico de 5’6 ppm. Los espectros de 1H-RMN de los diferentes productos se pueden consultar en el anexo 2. Los espectros de 13C presentan dos señales alrededor de 164 y 165 ppm, típicas del anillo de triazina, que en algunos casos eran difíciles de ver, debido a su poca intensidad. Los espectros de 13 C-RMN de los diferentes productos se encuentran en el anexo 2. En los espectros IR observamos la presencia de bandas correspondientes a la vibración de tensión de enlaces N-H en la zona próxima a 3400 cm-1. También podemos apreciar las vibraciones de tensión de los enlaces C=C y C=N entre 1450 y 1600 cm-1, aunque son bandas intensas difíciles de asignar y una banda en torno a 800 cm-1, que corresponde con bandas de deformación del anillo de triazina.82 En lo que respecta a los puntos de fusión, por lo general todas las bistriazinas se mantienen en estado sólido hasta altas temperaturas (unos 300 ºC), lo que da idea de la alta estabilidad de estos compuestos. Las únicas excepciones son las bistriazinas que presentan anillos aromáticos sin sustituir ya que, en el caso de la bistriazina 6b, el punto de fusión se sitúa sobre 160 ºC y, para 6f, en el entorno de 200 ºC. Para la monotriazina 5d tenemos un intervalo de fusión de 182-183 ºC. Los espectros de masas confirman la existencia del ion molecular M+H+. 2.3.2.3. Estudio de las propiedades ópticas: 2.3.2.3.1. Espectroscopia UV-visible: El estudio de las propiedades ópticas de los derivados de triazina sintetizados se llevó a cabo mediante espectroscopia UV-visible y fluorescencia a temperatura ambiente, siguiendo el protocolo descrito en la parte experimental para las medidas en 82 R. M. Desai, D. K. Dodiya, A. R. Trivedi, V. H. Shah, Med. Chem. Res., 2008, 17, 495-506. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 85 disolución de diclorometano. Los espectros de absorción normalizados se representan en la figura 2.21. 7 -Cl 6d Piperidina 6e Morfolina 6h p-NO2PhNH 1,0 1,0 Absorbancia Absorbancia 0,8 0,6 0,4 7 -Cl 6b p-OMePhNH 6c PhNH 6f Ph2N 0,5 0,2 0,0 0,0 300 400 500 600 Longitud de onda (nm) 250 300 350 400 Longitud de onda (nm) Figura 2.21. Espectros UV normalizados de las bistriazinas descritas en este trabajo (CH2Cl2, 10-5 M). Los resultados obtenidos se recogen en la tabla 2.12, y muestran que los máximos de absorción se encuentran en la región del UV, alrededor de 300 nm. Este hecho, junto con la alta absortividad molar se atribuye a transiciones π-π*.69 Tomando como referencia el derivado 7 (entrada 7, tabla 2.12), con cuatro átomos de cloro, la presencia de un grupo nitro, electroatractor, en la estructura del compuesto provoca un desplazamiento del segundo máximo de absorción a 355 nm, incrementando también su absorbancia. Por otro lado, cuando tenemos sustituyentes dadores en sistemas conjugados (casos de 6b, 6c y 6f) se observa un desplazamiento al rojo de los máximos de menor longitud de onda,40 si bien este desplazamiento es menor en el caso de 6f, debido a que esta molécula no es plana (entrada 5, tabla 2.12), como se muestra en la figura 2.22, lo que le hace perder parte de su conjugación. E. Beltrán, J. L. Serrano, T. Sierra, R. Giménez, Org. Let.. 2010, 12, 1404. M. M. Rothmann, S. Haneder, E. Da Como, C. Lennartz, C. Schildknetch, P. Strohriegl, Chem. Mater. 2010, 22, 2403. 69 40 86 2.3. Discusión de resultados Tabla 2.12 Entrada Compuestoa 1 6b R λabs (nm) [log ε] 269 [5’20], 323 [4’57] 2 6c 272 [5’02] 3 6d 237 [5’01], 336[4’28] 4 6e 236 [5’03], 336 [4’34] 5 6f 244 [4’97], 336 [4’49] 6 6h 238 [4’36], 355 [4’46] 7 7 -Cl 233 [4’60], 311 [4’08] -5 a) Disoluciones 10 M en CH2Cl2. 6b 6f Figura 2.22. Representaciones tridimensionales de las estructuras de los compuestos 6b (superior) y 6f (inferior). 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 2.3.2.3.2. 87 Fluorescencia: En primer lugar, compararemos las propiedades fluorescentes de la monotriazina 5d y la bistriazina 6d sintetizadas en este capítulo. Como cabría esperar, el aumento de la conjugación al pasar de una monotriazina a una bistriazina (tabla 2.13) provoca un incremento del rendimiento cuántico, que en nuestro caso es de 43 veces. Esta es la principal razón por la que nos hemos centrado en sintetizar y estudiar dichas bistriazinas. Tabla 2.13. Comparación de las propiedades ópticas de la monotriazina 5d y la bistriazina 6d. Compuesto λabs (nm) λfluoresc (nm) Desplazamiento ΦF -1 [log ε] de Stokes (cm ) 234 [4’73], 335 [4’05] 428, 400 19553 4673 237 [5’00], 336 [4’29] 389 16487, 4054 0’0069 0’30 A modo de ejemplo, en la figura 2.23 se muestran los espectros de UV y fluorescencia realizados al compuesto 6c. El resto de las gráficas se pueden ver en el anexo de esta memoria. En la tabla 2.14 se exponen los resultados obtenidos en estas experiencias. 88 2.3. Discusión de resultados UV Fluorescence 0,6 0,4 Intensity (a.u.) Absorbance 0,5 0,3 0,2 0,1 0,0 0 300 400 500 Wavelength (nm) Figura 2.23. Espectros UV y fluorescencia de 6c (CH2Cl2 10-5 M). Tabla 2.14. Propiedades ópticas de las 1,5-diaminonaftalenobistriazinas 6. Compuestoa R 6b λabs (nm) [log ε] 269 [5,20], 323 [4,57] λfluoresc (nm) Desplazamiento de Stokes (cm-1) 387 11334, 5119 ΦF 0’87 6c 272 [5,02] 387 10924 0’02 6d 237 [5’00], 336 [4’29] 389 16487, 4054 0’30 6e 236 [5,03], 336 [4,34] 387 16466, 3922 0’61 6f 244 [4,97], 336 [4,49] 375 390 15342, 4120 0’05 6h 238 [4,36], 355 [4,46] 387 16177, 2329 0’02 233 [4,60], 311 [4,08] 420 19108, 8401 0’0002 7 -Cl De los datos sobre los experimentos de fluorescencia que podemos ver en esta tabla pueden sacarse varias conclusiones: 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 89 1. Los máximos de fluorescencia prácticamente coinciden en todos los casos, salvo en el compuesto de partida tetraclorado 7, que está más desplazado hacia el rojo. 2. Los compuestos que tienen más de un máximo en UV (todos menos 6c) tienen sólo un máximo en fluorescencia, lo que quiere decir que los estados excitados a los que llega la molécula tras la absorción de luz están relacionados mediante procesos de pérdida de energía no radiativos. 3. Los desplazamientos de Stokes son muy elevados para las bandas de absorción cuyos máximos se encuentran por debajo de 300 nm lo que podría indicar la existencia de un sistema de separación de cargas. Por otro lado, en los máximos de absorción que sobrepasan los 300 nm los desplazamientos de Stokes tienen valores más bajos. 4. Cabe destacar el elevado rendimiento cuántico de 6b, próximo a 0’9. Este hecho es importante de cara a una posible aplicación en la fabricación de dispositivos optoelectrónicos, como OLEDs. Asimismo, el derivado de morfolina 6e alcanza un valor de 0’61. 5. Los máximos de fluorescencia se encuentran en la zona fronteriza entre el visible y el ultravioleta. También hay que destacar de estos espectros de fluorescencia que no se pudo realizar el estudio a diferentes concentraciones debido a que, al concentrar las disoluciones, la intensidad de fluorescencia aumenta hasta tal punto que se satura el espectrofluorímetro. Por ello, no podemos descartar la existencia de excímeros. 2.3.2.3.3. Excitación: Cuando los espectros de absorción y excitación no son superponibles, es indicativo de la existencia de varias especies en disolución o que una sola especie presente diferentes formas en el estado fundamental (como, por ejemplo, agregados). En la tabla 2.15 se ilustran los datos correspondientes a estas experiencias: 90 2.3. Discusión de resultados Tabla 2.15. Longitudes de onda de los máximos de cada espectro. Compuesto R λabs (nm) λfluoresc (nm) Máximos excitación (nm) 6b 269 323 387 277, 330 6c 272 387 279, 346, 365 6d 237 336 389 249, 340 6e 236 336 387 249, 338 6f 244 336 375 390 276, 337 6h 238 355 387 277, 344 233 311 420 330 7 -Cl 272, 348 En la tabla anterior tenemos diferentes casos, que ilustraremos con los correspondientes espectros (figuras de 2.24 a 2.26). En el caso del derivado 6b (figura 2.24) se puede observar que los máximos de los espectros de excitación y UV aparecen a unas longitudes de onda próximas. La diferencia que se aprecia en los máximos de longitud de onda inferior a 300 nm puede explicarse, al menos en parte, por la mayor absorción del disolvente en el espectro de UV, parámetro que no influye sobre el espectro de excitación. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 91 200 1,0 UV Excitacion 0,8 180 160 120 0,6 100 80 0,4 60 Intensidad (a.u.) Absorbancia 140 40 0,2 20 0,0 250 300 350 0 400 Longitud de onda (nm) Figura 2.24. UV y excitación de 6b (CH2Cl2, 10-5 M). En primer lugar, para el derivado clorado de partida 7, se observa que el espectro de excitación es totalmente diferente al de absorción (figura 2.25), lo que nos da idea de que existe más de una especie en la disolución. También es el caso de 6h y 6c, cuyos espectros pueden verse en el anexo 2. 20 UV Excitation 0,4 15 10 0,2 Intensity (a.u.) Absorbance 0,3 5 0,1 0,0 300 0 400 Wavelength (nm) Figura 2.25. Espectros UV y excitación del compuesto 7 (CH2Cl2, 10-5 M). 92 2.3. Discusión de resultados UV 350 Excitacion 1,0 300 0,8 0,6 200 150 0,4 Intensidad (u.a.) Absorbancia 250 100 0,2 50 0,0 250 300 350 0 400 Longitud de onda (nm) Figura 2.26. UV y excitación de 6d (CH2Cl2, 10-5 M). En el caso del derivado de piperidina 6d (figura 2.26), aunque parece que hay desplazamiento de 249 nm a 237 nm del máximo de la banda de excitación al de la banda de absorción, el hecho de que el máximo del espectro de absorción esté tan desplazado hacia el azul provoca que la absorción del disolvente interfiera en la medida, lo que hace muy difícil comprobar la identidad entre el espectro de ultravioleta y el de excitación. No obstante, la banda pequeña alrededor de 390 nm del espectro de excitación podría indicar la existencia de agregados, como comprobamos con estudios posteriores. 2.3.2.3.4. Influencia del disolvente: Realizamos un experimento de solvatocromismo a nuestros productos, estudiando la influencia del disolvente en las propiedades ópticas. Se examinan todas las bistriazinas, eligiendo diferentes tipos de disolvente, como hexano, diclorometano, acetonitrilo y metanol. Los resultados obtenidos se recogen en la tabla 2.16. Estos resultados indican la existencia de solvatocromismo, que en este caso sería positivo (desplazamiento batocrómico), ya que con disolventes más polares el máximo se desplaza hacia el rojo. De entre todos estos datos, el que más destaca es el correspondiente a 6c, donde los máximos en hexano no tienen nada que ver con los que podemos observar en el resto de disolventes (figura 2.27). 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 93 Tabla 2.16. Máximos UV en diferentes disolventes. Compuesto R 6b Hexano DCM CH3CN MeOH 212 234 269 269 268 343 323 318 317 6c 237 342 272 272 214 232 272 6d 234 332 237 336 234 333 233 329 6e 234 347 236 336 232 321 231 320 6f 210 234 343 244 336 239 242 334 6h 232 343 238 355 231 365 230 367 - 233 311 223 298 221 304 7 -Cl UV Hexane UV DCM UV CH3CN UV MeOH Absorbance 1,0 0,5 0,0 200 300 400 Wavelength (nm) Figura 2.27. Espectros de absorción normalizados de 6c en diferentes disolventes. Por otro lado, también se estudió la influencia del disolvente en los espectros de fluorescencia. Los datos obtenidos se muestran en la tabla 2.17. 94 2.3. Discusión de resultados Tabla 2.17. Máximos de intensidad de fluorescencia con diferentes disolventes. Compuesto R Hexano DCM CH3CN MeOH 6b 322 367 382 387 386 385 6c 367 382 387 386 385 6d 367 383 396 386 385 6e 367 382 386 386 387 409 6f 291 367 383 375 390 386 396 412 6h 367 383 387 436 387 385 413 - 420 384 394 7 -Cl Las diferencias existentes en los máximos de emisión con disolventes polares no son grandes. No se observa una tendencia lo suficientemente clara como para afirmar que existe un solvatocromismo. En el caso del diclorometano para el derivado 6h (figura 2.28), que presenta un máximo a 436 nm, muy diferente del resto de espectros, podemos pensar que esta banda intensa puede corresponder a la existencia de excímeros en disolución, lo que podría explicar su desplazamiento hacia el rojo. En general, a longitudes de onda más altas, al aumentar la polaridad del disolvente se produce un aumento de la intensidad de fluorescencia relativa. En resumen, aunque en algunos casos el desplazamiento de Stokes es elevado (tabla 2.14), lo que podría indicar procesos de transferencia de carga, las pruebas de influencia del disolvente no confirman esta hipótesis. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 95 Fluorescence Hexane Fluorescence DCM Fluorescence CH3CN Fluorescence MeOH 1,0 Intensity (a.u.) 0,8 0,6 0,4 0,2 0,0 300 400 500 Wavelength (nm) Figura 2.28. Estudio de solvatocromismo para el derivado 6h. 2.3.2.3.5. Estudios de agregación: Como se mencionó en la introducción de esta memoria, los derivados de 1,3,5triazina son susceptibles de experimentar interacciones supramoleculares de muy diversos tipos. Estas interacciones pueden originar la aparición de agregados. Los datos obtenidos en experiencias anteriores nos motivan a realizar estudios para comprobar si los productos experimentan agregaciones en disolución. Conviene destacar que los disolventes elegidos en cada caso van en función de la solubilidad de los productos y la concentración requerida para cada experiencia. Se han realizado varios tipos de experimentos: a) Método de encapsulación con Rojo Nilo.76 b) DLS (Dynamic Light Scattering). c) DOSY (Difussion Ordered SpectroscopY). 76 (a)Y. B. Lim, E. Lee, M. Lee, Angew. Chem. Int. Ed. 2007, 46, 9011-9014. (b) M. C. A. Stuart, J. C. van de Pas, J. Engberts, J. Phys. Org. Chem. 2005, 18, 929-934. 96 - 2.3. Discusión de resultados Método de encapsulación con Rojo Nilo: Llevamos a cabo el método de encapsulación con Rojo Nilo. Si nuestras bistriazinas agregan, dichos agregados provocarían la encapsulación de la molécula de Rojo Nilo, haciendo que la fluorescencia disminuya. La bistriazina con sustituyente fenilo 6b se eligió como patrón para comenzar los estudios de encapsulación con Rojo Nilo. El estudio consiste en realizar medidas de fluorescencia de disoluciones de nuestro producto a diferentes concentraciones, adicionando a éstas una concentración constante de Rojo Nilo. En las disoluciones más diluidas, de concentración inferior a la CAC,83 observamos que la intensidad de fluorescencia de la disolución es similar a la del blanco mientras que, cuando alcanzamos la concentración crítica de agregación, observamos que la intensidad de fluorescencia empieza a disminuir. Esto se debe a que tenemos menor concentración de Rojo Nilo libre en disolución, ya que ha empezado a intercalarse entre los agregados del producto. Para que estos experimentos puedan llevarse a cabo, es necesario que nuestros compuestos no emitan en la zona en la que lo hace la molécula de Rojo Nilo, condición que se cumple. Una vez obtenidos los datos, representamos en un gráfico el logaritmo de concentración de bistriazina respecto de la intensidad de fluorescencia (figura 2.29). En todos los casos se llevaron a cabo 3 medidas de cada muestra, tomando el valor medio como dato para la representación. En la gráfica se observan dos partes bien diferenciadas. En primer lugar, por debajo de la CAC, tenemos una intensidad de fluorescencia prácticamente constante e igual a la intensidad de fluorescencia del blanco (disolución patrón de Rojo Nilo). En segundo lugar, por encima de la CAC observamos cómo, al incrementarse la concentración de bistriazina, la intensidad de fluorescencia disminuye. Se realiza una recta de calibrado para cada parte de la gráfica, mediante el método de regresión por mínimos cuadrados, obteniendo el logaritmo de la CAC como la intersección de las dos 83 CAC (Concentración de agregación crítica). 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 97 rectas. El resultado es una concentración de agregación crítica de 1,04x10-5 M para el compuesto 6b en THF. 6b 170E+04 y = -13984x + 2E+06 R² = 0,2225 160E+04 150E+04 Intensidad (u.a.) 140E+04 130E+04 120E+04 110E+04 y = 177801x + 544269 R² = 0,8757 100E+04 3 4 5 6 7 8 -Log [C] Figura 2.29. Representación de –Log [6b] frente a la intensidad de fluorescencia (THF). En el caso de la bistriazina que tiene anillos de piperidina como sustituyentes (6d), la concentración de agregación crítica es casi dos órdenes de magnitud menor (3,75x10-7 M) que en el caso anterior, como podemos ver en la figura 2.30, es decir, tiene más tendencia a formar agregados que en el caso anterior en THF. 98 2.3. Discusión de resultados 6d 850000 y = 15401x + 595912 R² = 0,1721 y = 100390x + 49705 R² = 0,9306 750000 Intensidad (u.a.) 650000 550000 450000 350000 250000 3 4 5 6 7 8 9 Figura 2.30. Representación de –Log [6d] frente a la intensidad de fluorescencia (THF). - DLS: Se han realizado medidas de dispersión dinámica de la luz sobre la bistriazina 6b. Hemos obtenido la distribución de diámetro hidrodinámico de partículas que se muestra en la figura 2.31. La medida de DLS nos permite confirmar la existencia de agregados en la disolución analizada, con un diámetro hidrodinámico de 75 nm para la población mayoritaria. Los resultados obtenidos en esta experiencia concuerdan con los vistos para los ensayos de encapsulación de Rojo Nilo, que nos daba una concentración de agregación crítica de 1’04x10-5 M, ya que, en este caso, la concentración de producto usada para llevar a cabo las medidas de DLS (0’8 mM) es mayor que la concentración de agregación crítica. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 99 Normalized Counts 120 6b_800 uM_DCM 100 80 60 40 20 0 0 50 100 150 200 250 300 350 Hydrodynamic Diameter (nm) Figura 2.31. Distribución de radio hidrodinámico de una disolución de 6b a una concentración de 0’8 mM en CH2Cl2. También se han realizado medidas de DLS para otros derivados. Los datos obtenidos se representan en la tabla 2.18. Tabla 2.18. Resultados de los experimentos de DLS. Compuesto Concentración Disolvente ¿Forma Diámetro agregados? hidrodinámico 10-4 M THF Sí 88 nm 10-6 M THF Sí 128 nm 10-4 M THF Sí 97 nm 10-5 M THF Sí 107 nm 10-5 M THF Sí 115 nm 100 - 2.3. Discusión de resultados Medidas de DOSY: Las medidas DOSY proporcionan información de la distribución de tamaños de partícula que forman parte de la muestra, basándose en coeficientes de difusión de cada especie química en disolución. Dichos coeficientes dependen del tamaño y forma de las moléculas. Por desgracia, la baja solubilidad de las bistriazinas nos ha imposibilitado la preparación de muestras a las concentraciones requeridas para realizar dichos análisis. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 101 2.4. PARTE EXPERIMENTAL 2.4.1. EQUIPAMIENTO. Las reacciones se efectuaron en un horno microondas focalizado CEM DiscoverTM (figura 2.32) con medida y control de la temperatura mediante un lector infrarrojo. Posee un sistema de medida de presión y un sistema de enfriamiento para el control de la temperatura. Los distintos parámetros como potencia, temperatura y tiempo se pueden monitorizar, al igual que modificar mediante el transcurso de la reacción. Figura 2.32. Horno microondas focalizado CEM. Las cromatografías en capa fina se han realizado en cromatofolios de sílica gel F254 Merk de 0’2 mm de espesor, utilizando para su revelado una lámpara ultravioleta de 254 nm. Todos los productos de partida comerciales y disolventes se utilizaron sin previa purificación. Los espectros de resonancia magnética nuclear de 1H y 13C se han realizado en un espectrofotómetro Varian Inova-500 (figura 2.33) operando a 499’772 MHz para protón y 125’423 MHz para carbono-13. Los valores de desplazamiento químico (δ) se dan en partes por millón (ppm), utilizando tetrametilsilano como referencia interna y el disolvente indicado en cada caso. Las constantes de acoplamiento J se dan en Hz. 102 2.4. Parte experimental Figura 2.33. Espectrofotómetro Inova-500. Los espectros de infrarrojo se realizaron en un espectrofotómetro de infrarrojos de transformada de Fourier FT-IR Shimadzu IR Prestige-21 que incorpora un ATR (reflectancia total atenuada) con un objetivo de ZnSe (figura 2.34). Las medidas se hicieron con las muestras en estado sólido. Figura 2.34. Espectrofotómetro IR. Los espectros de masas se analizaron mediante la técnica de ionización de Electrospray (ESI), en el equipo QSTAR pulsar i de Applied Biosystems. Se realizaron registros en modo positivo, empleando ácido fórmico al 0’1 % en metanol como fase ionizante y aplicando una calibración externa para obtener los resultados en masa exacta. Los experimentos de UV-visible se realizaron utilizando un espectrofotómetro Jasco V-530, mientras que los espectros de fluorescencia fueron obtenidos empleando un espectrofluorímetro Jasco FP-750 (figura 2.35). Tanto en las medidas de absorción 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 103 UV-visible como en las medidas de fluorescencia en disolución se emplearon cubetas estándar de cuarzo de un centímetro de anchura. Figura 2.35. Espectrofotómetros de fluorescencia (izquierda) y UV-visible (derecha). Las disoluciones usadas para realizar los espectros de UV-visible y fluorescencia se prepararon por dilución a partir de disoluciones madre de concentración 10-3 M, para las cuales se pesaron las cantidades correspondientes de cada producto en la balanza analítica, y disolviendo el mismo en matraces aforados de 10 ml. La concentración de las disoluciones usadas, tanto para los experimentos de UV-visible como para los de fluorescencia, ha sido de 10-5 M. Los puntos de fusión fueron determinados mediante un lector de punto de fusión Büchi M-565 (figura 2.36). Figura 2.36. Lector de punto de fusión Büchi M-565. El diámetro hidrodinámico de las partículas fue determinado utilizando un instrumento de dispersión de luz dinámica (DLS, Zeta Plus, Brookhaven, Holstville, 104 2.4. Parte experimental NY), operando con un ángulo de dispersión de 90º a 635 nm y una fuente de diodo láser de 35 mW. Para estos estudios se usaban 3 ml de disolución de los compuestos en el disolvente apropiado a una concentración de 10-3 M, en el caso de que la solubilidad lo permitiese o, en su defecto, lo más concentrada posible sin tener turbidez. El camino óptico de la cubeta era de 1 cm. Cada muestra fue medida diez veces y cada medida duró 5 minutos a 298 K. Los datos fueron ajustados usando el algoritmo de mínimos cuadrados no restringidos negativamente para resolver la función de autocorrelación medida experimentalmente. 2.4.2. SÍNTESIS DE MONOTRIAZINAS CON 2,5DIMETOXIANILINA. En un matraz de microondas de boca B-14 esmerilada (figura 2.37), se introducen 2,5-dimetoxianilina y la clorotriazina66 correspondiente a cada producto en proporción 2:1. Figura 2.37. Matraz de microondas. Se homogeniza la mezcla y se introduce el matraz en el microondas durante 15 minutos a una temperatura de 150 ºC sin disolvente. Tras este proceso, se añaden 5 ml de CH2Cl2 a la mezcla de reacción y se realiza una filtración en una columna con gel de sílice (10 mm de altura por 12 mm de diámetro), y usando acetato de etilo como eluyente. El crudo de reacción se lava primero con 5 ml de HCl y seguidamente con 5 66 A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente. Org. Biomol. Chem. 2003, 1, 4451. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 105 ml de una disolución saturada de carbonato sódico. Los productos puros se obtienen por simple filtración. N-(2,5-Dimetoxifenil)-N’,N’’-bis-(2-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (3a). A partir de 6-cloro-N,N'-bis-(2-pirazol-1-ilfenil)-1,3,5-triazina-2,4-diamina (0’25 mmol, 0’110 g) y de 2,5-dimetoxianilina (0’5 mmol, 0’078 g). Se lava con 5 ml de HCl 1 M y, seguidamente, con 5 ml de una disolución saturada de carbonato sódico, obteniéndose 3a puro como un sólido blanco (0’126 g, 90 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’67 (s, 3H, OCH3-5); 3’79 (s, 3H, OCH3-2); 6’53 (d, J = 1’83 Hz, 2H, H4 pir); 6’59 (dd, J = 8’79 Hz y J = 2’93 Hz, 1H, H4 ArCH3); 6’94 (d, J = 8’79 Hz, 1H, H3 ArCH3); 7’23 (t, J = 7’69 Hz, 2H, H4 Ph); 7’35 (t, J = 7’87 Hz, 2H, H5 Ph); 7’53 (d, J = 7’6 Hz, 2H, H3 Ph); 7’69 (d, J = 2’56 Hz, H6 ArCH3); 7’81 (d, J = 1’1 Hz, 2H, H3 pir); 7’87 (s, 1H, NH); 8’15 (d, J = 2’56 Hz, 2H, H5 pir); 8’19 (d, J = 8’05 Hz, 2H, H6 Ph); 9’44 (s, 2H, NH). 13 C-RMN (DMSO, 80 ºC) δ: 56’30 (OMe-5) ; 57’21 (OMe-2); 106’65 (C4 pir); 107’83 (C4 ArCH3); 108’33 (C6 ArCH3); 111’71 (C3 ArCH3); 123’53 (C4 Ph); 123’55 (C3 Ph); 124’28 (C6 Ph); 127’07 (C5 Ph); 128’18 (C1 ArCH3); 130’66 (C2 Ph); 130’79 (C5 pir); 131’44 (C1 Ph); 140’47 (C3 pir); 143’54 (C2 ArCH3); 152’99 (C5 ArCH3); 163’71 (C6 Tz); 163’80 (C2,4 Tz). MS (ESI) M + H+: experimental: 547’2336; M teórica: 546’2240; 2M + H+: 1093’4601. Punto de fusión: 178-180 ºC, descompone. IR (Neto) υ (cm-1): 3398, 1575, 1506, 1417, 1217, 1049. N-(2,5-Dimetoxifenil)-N’,N’’-difenil-1,3,5-triazina-2,4,6-triamina (3b). A partir de 6-cloro-N,N'-difenil-1,3,5-triazina-2,4-diamina (0’25 mmol, 0’074 g) y de 2,5-dimetoxianilina (0’5 mmol, 0’078 g). Se lava con 5 ml de una disolución de 106 2.4. Parte experimental HCl 1 M y, seguidamente, con 5 ml de una disolución saturada de carbonato sódico obteniéndose 3b, que aparece como un sólido blanco puro (0’099 g, 95 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’71 (s, 3H, OCH3- 5); 3’83 (s, 3H, OCH3-2); 6’62 (d, J = 8’79 Hz, 1H, H4 ArCH3); 6’96 (d, J = 8’79 Hz, 1H, H3 ArCH3); 7’02 (t, J = 6’34 Hz, 2H, H4 Ph); 7’28 (t, J = 6’83 Hz, 4H, H3,5 Ph); 7’73 (d, J = 7’32 Hz, 4H, H2,6 Ph); 7’77 (s, 1H, NH); 7’83 (s, 1H, H6 ArCH3); 9’20 (s, 2H, NH). 13 C-RMN (DMSO, 80 ºC) δ: 56’33 (O OCH3-5); 57’26 (O OCH3-2); 108’20 (C4 ArCH3); 109’90 (C6 ArCH3); 112’66 (C3 ArCH3); 121’53 (C3,5 Ph); 123’10 (C4 Ph); 129’00 (C2,6 Ph); 129’49 (C1 ArCH3); 140’29 (C1 Ph); 144’57 (C2 ArCH3); 154’26 (C5 ArCH3); 164’47 (C6 Tz); 164’67 (C2,4 Tz). MS (ESI) M + H+: experimental: 415’1880; M teórica: 414’1804. Punto de fusión: 176-178 ºC, descompone. IR (Neto) υ (cm-1): 3392, 1514, 1417, 1398, 1230, 1049. N-(2,5-Dimetoxifenil)-N’,N’’-bis-(p-metoxifenil)-1,3,5-triazina-2,4,6-triamina (3c). A partir de 6-cloro-N,N'-bis-(p-metoxifenil)-1,3,5-triazina-2,4-diamina (0’25 mmol, 0’089 g) y de 2,5-dimetoxianilina (0’5 mmol, 0’078 g). Se lava con 5 ml de una disolución de HCl 1M y, acto seguido, con 5 ml de una disolución saturada de carbonato sódico obteniéndose 3c, que aparece puro como un sólido blanco (0’107 g, 90 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’69 (s, 3H, OCH3-5); 3’75 (s, 6H, OCH3(p)); 3’83 (s, 3H, OCH3-2); 6’56 (d, J = 8’78 Hz, 1H, H4 ArCH3); 6’86 (d, J = 8’78 Hz, 4H, H3,5 Ph-p-O OCH3); 6’93 (d, J = 8’78 Hz, 1H, H3 ArCH3); 7’47 (s, 1H, NH); 7’58 (d, J = 8’78 Hz, 4H, H2,6 Ph-p-O OCH3); 7’90 (s, 1H, H6 ArCH3); 8’90 (s, 2H, NH). 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 13 107 C-RMN (DMSO, 80 ºC) δ: 54’98 (OCH3-p); 55’19 (OCH3-5); 56’18 (OCH3-2); 106’30 (C4 ArCH3); 108’05 (C6 ArCH3); 111’32 (C3 ArCH3); 113’40 (C3,5 Ph-p-O OCH3); 122’18 (C2,6 Ph-p-O OCH3); 128’85 (C1 ArCH3); 132’46 (C1 Ph-p-O OCH3); 142’83 (C2 ArCH3); 153’19 (C5 ArCH3); 154’70 (C4 Ph-p-O OCH3); 163’74 (C6 Tz); 164’01 (C2,4 Tz). MS (ESI) M + H+: experimental: 475’2108; M teórica: 474’2016. Punto de fusión: 180-181 ºC, descompone. IR (Neto) υ (cm-1): 3419, 3394, 1512, 1409, 1236, 1026. N-(2,5-Dimetoxifenil)-N’,N’’-bispiperidino-1,3,5-triazina-2,4,6-triamina (3d). A partir de 6-cloro-N,N'-bis-piperidino-1,3,5-triazina-2,4-diamina (0’25 mmol, 0’070 g) y de 2,5-dimetoxianilina (0’5 mmol, 0’078 g). Se lava con 5 ml de una disolución 0’1 M de HCl y, posteriormente, con 5 ml de una disolución saturada de carbonato sódico, obteniéndose 3d puro como un sólido blanco (0’084 g, 85 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 1’56 (m, 8H, H3 y H5 piperidina), 1’65 (m, 4H, H4 piperidina), 3’71 (s, 3H, OCH3-5), 3’74 (t, J = 5’49 Hz, 8H, H2 y H6 piperidina), 3’82 (s, 3H, OCH3-2), 6’61 (dd, J = 9’15 Hz y J = 3’29 Hz, 1H, H4 Ph), 6’97 (d, J = 9’15 Hz, 1H, H3 Ph), 7’89 (d, J = 3’29 Hz, 1H, H6 Ph), 8’55 (s, 1H, NH) 13 C-RMN (DMSO, 80 ºC) δ: 23’59 (C4 piperidina), 24’92 (C3 y C5 piperidina), 44’30 (C2 y C6 piperidina), 55’18 (OCH3-5), 56’43 (OCH3-2), 106’73 (C6 Ph), 108’35 (C4 Ph), 112’16 (C3 Ph), 127’71 (C1 Ph), 142’97 (C2 Ph), 152’98 (C5 Ph). MS (ESI) M + H+: experimental: 399’2492; M teórica: 398’2430. Punto de fusión: 202-206 ºC, descompone. IR (Neto) υ (cm-1): 2933, 2848, 1589, 1504, 1022. N-(2,5-Dimetoxifenil)-N’,N’’-bismorfolino-1,3,5-triazina-2,4,6-triamina (3e). A partir de 6-cloro-N,N'-bis-morfolino-1,3,5-triazina-2,4-diamina (0’25 mmol, 0’071 g) y de 2,5-dimetoxianilina (0’5 mmol, 0’078 g). Se lava con una disolución 0’1 M de HCl y, después, con 5 ml de una disolución saturada de carbonato sódico para obtener 3e puro como un sólido blanco (0’080 g, 80 %). 108 2.4. Parte experimental Datos físicos y espectroscópicos: 1 H-RMN (DMSO, TA) δ: 3’61 (m, 8H, H3,5 Morfolina), 3’69 (m, 11H, OCH3-5, H2,6 Morfolina), 3’80 (s, 3H, OCH3-2), 6’51 (dd, J = 2’93 Hz y J = 8’78 Hz, 1H, H4 Ph), 6’92 (d, J = 8’78 Hz, 1H, H3 Ph), 7’48 (s, 1H, NH), 7’92 (d, J = 3’42 Hz, 1H, H6 Ph). 13 C-RMN (DMSO, TA) δ: 43’32 (C2,6 Morfolina), 55’16 (OCH3(5)), 56’23 (OCH3(2)), 65’93 (C3,5 Morfolina), 106’10 (C6 Ph), 106’46 (C4 Ph), 111’38 (C3 Ph), 129’04 (C1 Ph), 142’30 (C2 Ph), 152’95 (C5 Ph), 163’63 (C6 Tz), 164’57 (C2,4 Tz). MS (ESI) M + H+: experimental: 403’2092; M teórica: 402’2016. Punto de fusión: 201-206 ºC, descompone. IR (Neto) υ (cm-1): 3414, 1575, 1506, 1255, 1114, 1043. 2.4.3. SÍNTESIS DE DERIVADOS DE TRIAZINA CON 1,5DIAMINONAFTALENO. N1-(4,6 Di(piperidino)-1,3,5-triazin-2-il)naftaleno-1,5-diamina (5d). En un matraz microondas de boca B-14 se introduce 2-cloro-4,6-di(piperidino)1,3,5-triazina (0’5 mmol, 0’140 g) y 1,5-diaminonaftaleno (1’5 mmol, 0’237 g). La mezcla se homogeniza, se introduce en un horno microondas y se irradia a 180°C durante 25 minutos. El crudo de reacción se disuelve en 5 ml de acetona. A continuación, se añaden 0’6 mL de HCl 0’1 M y se filtra a través de un filtro de pliegues. Al filtrado se le añaden 0’5 mL de HCl 1M, precipitando un sólido que se separa por filtración. A este nuevo filtrado se le añaden unos 5 mL de H2O precipitando un sólido que se suma al anterior (0’125g, 62%). Datos físicos y espectroscópicos 1 H-RMN (DMSO) TA: 1’44 (s, 8H, H3,5 piperidina), 1’57 (d, J = 4’8 Hz, 4H, H4 piperidina), 3’62 (s, 8H, H2,6 piperidina), 5’63 (s, 2H, NH2), 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 109 6’66 (d, J = 7’7 Hz, 1H, H6), 7’15 (t, J = 7,3 Hz, 1H, H7), 7’23 (d, J = 8’4 Hz, 1H, H8), 7’31 (t, J = 7’7, 1H, H3), 7’64 (d, J = 7’3, 1H, H2), 7’82 (d, J = 8’4, 1H, H4), 8’54 (s ancho, 1H, NH) 13 C-RMN (DMSO) TA: 24’4 (C4 piperidina), 25’4 (C3,5 piperidina), 43’4 (C2,6 piperidina), 107’4 (C6), 110’7 (C8), 118’3 (C4), 121’3 (C2), 122’8 (C3), 123’4 (C4a), 126 (C7), 129’5 (C8a), 135 (C1), 144’7 (C5), 164’6 (C4,6Tz), 165’4 (C2Tz). MS (ESI) M + H+: experimental: 404’2565, M teórica: 403’2486. Punto de fusión: [182-183] ºC. IR (Neto) ν (cm-1): 3346, 2927, 2846, 1512, 1485. Para sintetizar las bistriazinas, se introduce una mezcla de N1,N5-bis(4,6-dicloro1,3,5-triazin-2-il)naftaleno-1,5-diamina (7)81 (0’25 mmol, 0’113 g) con 2 mmol de la amina o anilina correspondiente en un matraz de microondas. La mezcla se homogeniza y se somete a radiación microondas durante el tiempo adecuado. Se adiciona una disolución de HCl 0’1 M (5 ml) al crudo de reacción, que es sonicado, filtrado y lavado con 1 ml de diclorometano. El sólido resultante es la bistriazina pura. N2,N2'-(Naftaleno-1,5-diil)bis(N4,N6-difenil-1,3,5-triazina-2,4,6-triamina) (6b). A partir de anilina (2 mmol, 0’186 g). Se somete la mezcla a radiación microondas durante 10 minutos a 140 ºC, obteniendo un rendimiento de 0’166 g (98 %). Datos físicos y espectroscópicos: 1 H RMN (DMSO, 80 ºC) δ: 6’95 (s, 4H, H4 Ph); 7’21 (s, 8H, H3 and H5 Ph); 7’57 (d, 2H, J = 7’81 Hz, H3 and H7 Naph.); 7’71 (d, 10 H, J = 6’25 Hz, H2 and H6 Ph, H4 and H8 Naph.); 8’00 (d, 2H, J = 7’81 Hz, H2 and H6 Naph.); 8’96 (s, 4H, NH Ph); 9’15 (s, 2H, NH Naph.). 13 C-RMN (DMSO, 80 ºC) δ: 120’0 (C2,6 Ph); 120’6 (C2,6 Naph.); 121’6 (C4 Ph); 123’6 (C4,8 Naph.); 124’9 (C3,7 Naph.); 127’8 (C3,5 Ph); 130’3 (C4a,8a Naph.); 134’6 (C1,5 Naph.); 139’5 (C1 Ph); 163’9 (C4,6 Tz); 165’5 (C2 Tz). MS (FAB) m/z 681’2 [M+H] +; HRMS calculado para C40H32N12 m/z: 680’2858, encontrado 680’2873. 81 W. Wei, H-J. Wang, C-Q. Jiang, Spectrochimica Acta Part A, 2008, 70, 362-366. 110 2.4. Parte experimental Punto de fusión: 155-156 ºC. IR (Neto) υ (cm-1): 3439, 1440, 1392, 1228, 806. N2,N2'-(Naftaleno-1,5-diil)bis(N4,N6-bis(4-metoxifenil)-1,3,5-triazina-2,4,6triamina) (6c). A partir de p-anisidina (2 mmol, 0’246 g). Se somete la mezcla a radiación microondas durante 10 minutos a 140 ºC, obteniendo un rendimiento del 65 % (0’130 g). Datos físicos y espectroscópicos: 1 H RMN (DMSO, 80 ºC) δ: 3’71 (s, 12H, CH3); 6’78 (d, 8H, J = 8’79 Hz, H3,5 Ph); 7’54 (m, 10H, H3,7 Naph. and H2,6 Ph); 7’69 (d, 2H, J = 7’32 Hz, H4,8 Naph.); 7’96 (d, 2H, J = 8’30 Hz, H2,6 Naph.); 8’80 (s, 4H, NH Ph); 9’06 (s, 2H, NH Naph.). 13 C-RMN (DMSO, 80 ºC) δ: 54’9 (OCH3); 113’3 (C3,5 Ph); 120’4 (C2,6 Naph.); 121’9 (C2,6 Ph); 123’3 (C4,8 Naph.); 124’9 (C3,7 Naph.); 130’1 (C1,5 Naph.); 132’4 (C1 Ph); 134’5 (C4a,8a Naph.); 154’6 (C4 Ph); 163’2 (C4,6 Tz); 164’8 (C2 Tz). MS (FAB): m/z 801’6 [M+H]+; HRMS calculado para C44H40N12O4 m/z: 800’3374, encontrado 800’3346. Punto de fusión: 288-292 ºC descompone. IR (Neto) υ (cm-1): 3406, 3342, 1487, 1392, 1232, 825. N1,N5-Bis(4,6-di(piperidin-1-il)-1,3,5-triazin-2-il)naftaleno-1,5-diamina (6d). A partir de 1,5-diaminonaftaleno (0’5 mmol, 0’079 g) y 2-cloro-4,6-di(piperidin-1-il)1,3,5-triazina66 (1’1 mmol, 0’308 g). Se homogeniza la mezcla y se somete a radiación microondas durante una hora a 200 ºC. Se añade acetona (5 ml) al crudo de reacción, que es filtrado para obtener el producto, con un rendimiento de 0.235 g (72 %). 66 A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. De la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente, Org. Biomol. Chem., 2003, 1, 4451. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 111 Datos físicos y espectroscópicos: 1 H-RMN (DMSO, TA) δ: 1’46 (s, 16H, H3,5 piperidina); 1’60 (s, 8H, H4 piperidina); 3’65 (s, 16H, H2,6 piperidina); 7’41 (t, J = 7’8 Hz, 2H, H3,7 Naph.); 7’69 (d, J = 7’8 Hz, 2H, H2,6 Naph.); 7’82 (d, J = 8’3 Hz, 2H, H4,8 Naph.); 8’56 (s, 2H, NH). 13 C-RMN (DMSO, 80 ºC) δ: 21’5 (C4 pip); 24’0 (C3,5 pip); 43’7 (C2,6 pip); 119’1 (C4,8 Naph.); 120’9 (C2,6 Naph.); 124’1 (C3,7 Naph.); 128’9 (C1,5 Naph.); 164’5 (C4,6 Tz.); 165’1 (C2 Tz). MS (FAB): m/z 649’4 [M+H] +; HRMS calculado para C36H48N12 648’4125, encontrado 648’4178. Punto de fusión: 247-252 ºC, cambio de fase. IR (Neto) υ (cm-1): 3464, 2929, 2848, 1593, 1514. N1,N5-Bis(4,6-dimorfolino-1,3,5-triazin-2-il)naftaleno-1,5-diamina (6e). A partir de morfolina (2 mmol, 0’174 g). Se somete la mezcla a irradiación microondas durante 10 minutos a 120 ºC. Se obtiene un rendimiento de 0’140 g (85 %). Datos físicos y espectroscópicos: 1 H RMN (CDCl3, TA) δ: 3’72 (s, 8H, H3 and H5 Morfolina); 3’77 (s, 8H, H2 and H6 Morfolina); 7’50 (dd, 2H, J = 7’8 Hz, J = 8’3 Hz, H3 and H7 Naph.); 7’81 (d, 2H, J = 8’79 Hz); 8’05 (d, 2H, J = 7’3 Hz). 13 C-RMN (CDCl3, TA) δ: 43’72 (C2,6 Morfolina); 66’82 (C3,5 Morfolina); 117’25 (C4,8 Naph.); 119’70 (C2,6 Naph.); 125’43 (C3,7 Naph.); 128’02 (C1,5 Naph); 134’54 (C4a,8a Naph.); 165 (C2 Tz). MS (FAB) m/z 657’5 [M+H] +; HRMS calculado para C32H40N12O4 656’3295, encontrado 656’3300. Punto de fusión: 332-335 cambio de fase. IR (Neto) υ (cm-1): 3464, 2980, 2900, 1516, 1253, 1118. N2,N2'-(Naftaleno-1,5-diil)bis(N4,N4,N6,N6-tetrafenil-1,3,5-triazina-2,4,6-triamina) (6f). 112 2.4. Parte experimental A partir de difenilamina (2 mmol, 0’338 g). Se somete la mezcla a radiación microondas durante 10 minutos a 200 ºC. Se obtiene un rendimiento de 0.162 g (65 %). Datos físicos y espectroscópicos: 1 H RMN (DMSO, 80 ºC) δ: 7’08-7’21 (m, 42 H, H2, H3, H4, H5 and H6 Ph, H3 and H7 Naph.); 7’42 (d, 2H, J = 7’32 Hz, H2 and H6 Naph.); 7’66 (d, 2H, J = 8’30 Hz, H4 and H8 Naph.); 8’98 (s, 2H, NH). 13 C-RMN (DMSO, 80 ºC) δ: 118’97; 121’36; 124’06; 124’78; 127’21; 128’04; 128’41; 128’93; 133’76; 141’69; 142’64; 143’11; 151’20; 164’91 (C2 Tz). MS (FAB) m/z 985’5 [M+H] +; HRMS calculado para C64H48N12 984’4125, encontrado 984’4114. Punto de fusión: 201-202 ºC. IR (Neto) υ (cm-1): 3230, 3095, 1546, 1236. N2,N2'-(Naftaleno-1,5-diil)bis(N4,N6-bis(4-nitrofenil)-1,3,5-triazina-2,4,6-triamina) (6h). A partir de 4-nitroanilina (2 mmol, 0’276 g). Se somete la mezcla a radiación microondas durante 10 minutos a 150 ºC. Se obtiene un rendimiento de 0’120 g (56 %). O2N Datos físicos y espectroscópicos: NH O2N N N H 1 N N H RMN (DMSO, 80 ºC) δ: 7’62 (t, 2H, J = 7’8 Hz, H3 and H7 Naph.); 7’67 (d, 2H, J = 6’8 Hz, H4 and H8 NH Naph.); 7’83 (s ancho, 8H, H2 and H6 Ph); 7’91 (s HN H N N N ancho, 8H, H3 and H5 Ph); 8’06 (d, 2H, J = 8’3 Hz, H2 N NO2 HN NO2 13 and H6 Naph.); 9’67 (s, 2H, NH Naph.); 9’77 (s, 4H, NH Ph). C-RMN (DMSO, 80 ºC) δ: 118’7 (C2,6 Ph); 121’7 (C2,6 Naph.); 123’8 (C3,5 Ph and C4,8 Naph.); 125’1 (C3,7 Naph.); 130’5 (C4a,8a Naph.); 134’5 (C1,5 Naph.); 140’7 (C4 Ph); 146’0 (C1 Ph); 163’8 (C4,6 Tz); 165’9 (C2 Tz). MS (ESI+) m/z 861’2 [M+H] +; HRMS calculado para C40H29N16O8 861’2348, encontrado 861’2361. Punto de fusión: 340-345 ºC cambio de fase. IR (Neto) υ (cm-1): 3327, 1504, 1323, 1111, 798. 2. Síntesis de mono y bistriazinas. Estudio de sus propiedades ópticas. 113 2.5. CONCLUSIONES Empleando la radiación microondas como fuente de energía, hemos logrado sintetizar nuevos derivados de 1,3,5-triazina, siguiendo una metodología medioambientalmente benigna, con tiempos cortos de reacción, sin disolvente y con una etapa de purificación simple. Esto puede aplicarse tanto a las 2,5- dimetoxifenilaminotriazinas como a las bistriazinas con puente 1,5-diaminonaftaleno. Las monotriazinas con sustituyentes 2,5-dimetoxifenilamina se han obtenido con rendimientos comprendidos entre el 85 y el 95 %. Los espectros de fluorescencia de estos derivados muestran la formación de excímeros. Y las medidas de Dynamic Light Scattering (DLS) confirman la formación de agregados con diámetros hidrodinámicos de hasta 296 nm. Los rendimientos cuánticos de fluorescencia de estos compuestos son bajos y, por tanto, no son útiles para fabricar OLEDs. Los derivados de 1,5-diaminonaftilbistriazinas también se han obtenido con altos rendimientos. Cabe destacar el excelente rendimiento cuántico de la bistriazina con sustituyente fenilo 6b, próximo a 0’9. Este hecho es importante de cara a una posible aplicación en la fabricación de dispositivos optoelectrónicos, como OLEDs. Asimismo, el derivado de morfolina 6e alcanza un valor de 0’61. Estudios de encapsulación con Rojo Nilo hacen posible calcular la concentración de agregación crítica (CAC) de los derivados de fenilo y piperidina que son respectivamente de 1,04 x 10-5 M y 3,75 x 10-7 M. Por otro lado, las medidas de DLS muestran que la mayor parte de las bistriazinas son capaces de formar agregados a concentraciones tan bajas como 10-5 M, hecho que confirma los datos de CAC obtenidos, lo que nos hace pensar que existe un gran número de interacciones supramoleculares. 114 2.4. Parte experimental 3. Síntesis de derivados de triazinilglicina. Sensores 3. SÍNTESIS DE DERIVADOS DE TRIAZINILGLICINA. SENSORES. 3.1. OBJETIVOS El objetivo principal de este capítulo es la síntesis de derivados de triazina con grupos aminoácido libres. Para ello, también nos proponemos como meta que el método sintético utilizado sea sostenible. Tanto los derivados de aminofeniltriazinas como los triazinilaminoácidos podrían ser anclados en nanoestructuras de carbono (figura 3.1) mediante la conocida reacción de Prato y, desde esta base carbonada, las interacciones supramoleculares que aportarían las triazinas podrían suponer una aplicación muy importante en reconocimiento molecular. Figura 3.1. Anclajes sobre nanoestructuras de carbono, concretamente sobre C60. Además del reconocimiento molecular, las interacciones supramoleculares podrían ser útiles en la formación de complejos con metales (figura 3.2), por lo que también nos proponemos como objetivo la obtención de complejos metálicos con nuestros derivados de triazinilglicina como ligando. Estos complejos formados podrían ser útiles para la detección de cationes, como Hg2+ o Zn2+, por lo que se explorará su potencial aplicación como sensores. 117 Figura 3.2. Puntos potenciales de coordinación de las triazinilglicinas. 3. Síntesis de derivados de triazinilglicina. Sensores 3.2. INTRODUCCIÓN 3.2.1. CATIONES METÁLICOS En esta sección hablaremos de los cationes metálicos que tienen importancia en este trabajo. Para ello, enmarcaremos a estos cationes en la naturaleza, en las aplicaciones que han tenido a lo largo de la historia, así como los efectos que pueden tener para la salud, tanto positivos como negativos. 3.2.1.1. Zinc:84 El Zn se caracteriza por ser un elemento ampliamente distribuido en la naturaleza, pero no es abundante, ya que representa sólo el 0,012% de la corteza terrestre. En el suelo, su concentración media es de 50 mg/kg. Actualmente, la mayor parte del zinc producido se emplea en la galvanización del hierro y acero, así como en la manufactura del latón. Los objetos galvanizados se emplean en la fabricación de una amplia diversidad de utensilios. También se utilizan grandes cantidades de zinc en la obtención de aleaciones, y en polvo se usa como agente reductor. Dentro de los compuestos, el óxido de zinc es el más importante, siendo empleado, entre otras cosas, como pigmento blanco. El Zn es uno de los elementos esenciales más abundantes en el cuerpo humano, encontrándose principalmente en el citosol de las células. Su cantidad en el individuo adulto oscila entre 1 y 2,5 g, siendo el segundo oligoelemento en relación a la cantidad total en el organismo, sólo superado por el hierro. Las concentraciones más elevadas aparecen en el hígado, páncreas, riñones, huesos y músculos voluntarios, existiendo también concentraciones importantes en el ojo, próstata, espermatozoides, piel, pelo y uñas. Para valorar su estatus en el organismo se usan principalmente como biomarcadores los niveles en suero, plasma y eritrocitos. C. Rubio, D. González Weller, R. E. Martín-Izquierdo, C. Revert, I. Rodríguez y A. Hardison, Nutr. Hosp., 2007, 22, 1, 101-107 y referencias allí citadas. 84 119 120 3.2. Introducción El Zn interviene en funciones fisiológicas necesarias para el desarrollo de la vida. Entre estas cabe destacar la función cerebral, crecimiento e integridad celular, visión nocturna o maduración sexual. 3.2.1.1.1. Fuentes dietéticas de zinc: El zinc está extensamente distribuido en alimentos y bebidas pero, tal como ocurre con otros elementos, los contenidos son tremendamente variables y, en general, bajos. Los productos de origen marino, principalmente los mariscos (ostras y crustáceos), son los alimentos más ricos en Zn, seguidos de las carnes rojas, derivados lácteos y huevos, y los cereales integrales. El zinc procedente de los alimentos vegetales es de menor biodisponibilidad, debido a la presencia de ácido fítico que forma complejos insolubles poco absorbibles. En los alimentos, el Zn se halla asociado particularmente a las proteínas y ácidos nucleicos, lo que va a condicionar en cierta medida su biodisponibilidad. En aguas de abastecimiento público, los contenidos en zinc pueden provenir, en parte, de la disolución de los terrenos y en parte de la cesión a partir de los materiales de las conducciones. En el anexo C de la Reglamentación Técnico-Sanitaria para el abastecimiento y control de las aguas potables de consumo público,85 se establece un valor guía de 100 µg/L de zinc, indicándose que a valores superiores a los 5 µg/L pueden aparecer sabores astringentes, opalescencias y depósitos granulosos. El procesado de alimentos es una de las principales causas de la pérdida de zinc. El ejemplo más representativo de este efecto lo constituyen los cereales, que pueden ver reducido su contenido desde un 20 a un 80% cuando son refinados. Por ello, se debe tener una especial consideración con las personas vegetarianas, ya que en estas personas los cereales son la principal fuente de zinc en la dieta. Si a la pérdida del 20-80% del contenido de zinc durante el refinado unimos que la biodisponibilidad del zinc en este tipo de dietas está disminuida si el contenido de fitato es alto, se concluye que la absorción y por tanto, la presencia de zinc en personas que siguen dietas vegetarianas es menor que en las que no las siguen. Real Decreto 1138/90, de 14 de septiembre, por el que se aprueba la Reglamentación Técnico-Sanitaria para el abastecimiento y control de las aguas potables de consumo público. BOE (226):27488-97. 85 3. Síntesis de derivados de triazinilglicina. Sensores 3.2.1.1.2. Ingesta dietética recomendada de zinc: Las recomendaciones de nutrientes (RDA = Recommended Dietary Allowance o IDR = Ingesta Diaria Recomendada) se definen como los niveles de ingesta de nutrientes considerados esenciales, según el criterio de los comités nacionales e internacionales que los establecen en base a los conocimientos científicos y que cubren las necesidades conocidas de prácticamente todas las personas sanas. Los valores de IDR se fijan en función de la edad, sexo, situación fisiológica (embarazo, lactancia, etc.) y normalmente son superiores a los verdaderos requerimientos. Las pérdidas endógenas en seres humanos oscilan entre los 1,3 y 4,6 mg/día. La ingesta recomendada de zinc para un adulto se sitúa entre 8 mg/día para las mujeres y 11 mg/día para los hombres. Durante la gestación y la lactancia las necesidades son ligeramente más altas. 3.2.1.1.3. Déficit de zinc: La deficiencia de este elemento en niños y jóvenes se debe a la escasez de alimentos de origen animal, dieta con un alto contenido en fitatos, inadecuada ingesta de alimentos y un incremento de las pérdidas fecales, pudiendo ocasionar retraso en el crecimiento y en el desarrollo neuronal, diarrea, alteraciones inmunitarias e incluso en algunos casos la muerte. Las manifestaciones principales son dermatitis, alopecia, alteraciones en el sentido del gusto, anorexia, retraso en la cicatrización de las heridas, alteraciones inmunológicas y disminución de los niveles de fosfatasas alcalinas, habiéndose postulado la deficiencia de zinc como un factor importante en la patogenia de la esquizofrenia. Alteraciones en la homeostasis del zinc se han relacionado con el Parkinson, el Alzheimer, isquemia cerebral transitoria, ataques de apoplejía, daños cerebrales e incluso un incremento en el riego de padecer cáncer. 3.2.1.1.4. Toxicidad del zinc: A pesar de que el zinc es el menos tóxico de todos los oligoelementos, y aunque su margen de seguridad (diferencia entre la dosis tóxica y la dosis recomendada) es muy 121 122 3.2. Introducción amplio, es necesario evaluar su toxicidad. Ello se puede establecer mediante el estudio de la Tolerable Upper Intake Level (UL), que se define como el nivel más alto de la ingesta diaria de un nutriente que no supone un riesgo o efectos adversos sobre la salud de casi todos los individuos. Este parámetro se calcula a partir de la ingesta total. Para el Zn proveniente tanto de los alimentos, como del agua y suplementos el UL es de 40 mg/día. Se ha demostrado que en hombres, un elevado consumo de suplementos de zinc produce un riesgo significativamente mayor de cáncer avanzado de próstata, así como la inhibición de los efectos beneficiosos de los biofosfonatos, el incremento de los niveles de testosterona, incremento de colesterol, reducción de los niveles de HDL (High Density Lipoprotein Cholesterol) y puede fomentar una disfunción inmune. Una suplementación con zinc, especialmente en altas dosis, también puede producir otros efectos adversos como interferir y disminuir el estatus corporal de cobre. Un caso especial se describe en un estudio realizado por Salzman y colaboradores86 en 2002 en el que los autores describen la intoxicación por zinc de un individuo de 17 años que durante 6-7 meses tomó elevadas dosis diarias de zinc en forma de suplementos y que desarrolló una hipocupremia con anemia, leucopenia y neutropenia. Esta anemia inducida por una hipocupremia por un exceso de zinc también, además de una nefrosis, se observa en otro caso de ingesta elevada de zinc (concretamente 2.000 mg de gluconato de zinc durante 12 meses). En ambos casos los efectos tóxicos remitieron al suprimir las ingestas de zinc. La inhalación de altas concentraciones de este metal, concretamente en forma de cloruro de zinc, puede causar neumonitis y un síndrome respiratorio en el adulto. In Vitro, el Zn produce citotoxicidad por una disminución de los niveles de glutatión reducido y un incremento de los niveles de la forma oxidada del glutatión. También in vitro, y a niveles elevados, produce muerte celular debido a que en primer lugar es capaz de generar especies reactivas de oxígeno y en segundo lugar a que activa la cascada de la MAP-kinasa. 86 M. B. Salzman, E. M. Smith, C. Koo, . J. Pediatr. Hematol. Oncol., 2002; 24, 7, 582-584. 3. Síntesis de derivados de triazinilglicina. Sensores 3.2.1.2. Mercurio:87 El mercurio y el cinabrio, principal mineral donde se obtiene, se conocen desde la antigüedad. Prueba de ello es que, en el inicio de sus culturas, pueblos como China, Egipto o Asiria conocían la existencia del cinabrio y lo aplicaban como pintura en forma de bermellón (polvo de cinabrio). Otras civilizaciones más modernas que emplearon el mercurio o el cinabrio fueron los fenicios (para extraer y purificar oro), los indios (creían que el mercurio era afrodisíaco), griegos, romanos o incas (estas tres como pintura, igual que en China o Egipto). Algunos médicos griegos, como Hipócrates, usaban el cinabrio como ungüento porque no lo consideraban tóxico por vía dérmica. La mina de cinabrio más grande del mundo, explotada desde la época romana hasta 2002, se encuentra en la cercana localidad de Almadén (Ciudad Real). Durante la Edad Media, época en la que la alquimia vivió su apogeo, el mercurio tuvo su importancia, si bien su consumo no aumentó hasta que aparecieron las primeras aplicaciones tecnológicas en la Edad Moderna, como el tratamiento de la sífilis (Paracelso, en el siglo XVI), el barómetro (Torricelli, en 1643) o el termómetro (Fahrenheit, en 1720). En la naturaleza, podemos encontrar el mercurio en forma de cinabrio, que es un sulfuro de mercurio o, principalmente, en grandes bolsas de mercurio metal. El sulfuro de mercurio es prácticamente inerte frente a los agentes atmosféricos (CO2, O2 y H2O) y no entra en el ciclo del agua, lo que hace despreciable su incorporación a las cadenas tróficas por esta vía. Así, esta incorporación ocurre a partir del propio Hg metal, ya que es volátil y a temperatura ambiente se está evaporando, con lo que se incorpora a la atmósfera en forma de vapor, sufriendo procesos posteriores de transformación en la especie soluble Hg2+. Hay que destacar que, dentro de las cadenas tróficas, el mercurio sufre procesos de bioconcentración, sobre todo en los animales marinos y los cereales, lo que hay que tener en cuenta como fuente de contaminación accidental. 87 X. Gaona Martínez, Tesis Doctoral. 2004. pp 9-49. Universidad Autónoma de Barcelona. 123 124 3.2. Introducción Además de la fuente natural de contaminación que ocurre con la evaporación del Hg metal existen, como suele ser habitual con los metales tóxicos, fuentes antropogénicas. Dentro de estas fuentes antropogénicas cabe destacar el uso del mercurio como fungicida, herbicida y conservante de semillas en agricultura, así como diferentes industrias, como la electroquímica. En la naturaleza, el mercurio experimenta un ciclo que resumen Mason y colaboradores88 con los diagramas de flujo que podemos ver en la figura 3.3 a y b, en el que se pueden apreciar las proporciones de mercurio en los diferentes medios y la influencia que la actividad humana ha tenido sobre este ciclo. En el agua, la especie predominante es la de Hg2+, muy soluble y que puede ser bioacumulado directamente por los peces, o puede ser biotransformado por microorganismos acuáticos, dando lugar a dos especies orgánicas: el dimetilmercurio, volátil, que se recicla a la atmósfera y el metilmercurio, que se bioacumula en los peces, lo que hace que se incorpore a las cadenas tróficas. También puede sufrir otras transformaciones. La toxicidad del mercurio es conocida desde la antigüedad. Hipócrates (370 a.C.) y Plinio (77 a.C.) describieron enfermedades y dolencias que sufrían los esclavos que trabajaban en las minas de mercurio. En los siglos XV y XVI empezaron a publicarse trabajos acerca de los efectos tóxicos del vapor de mercurio como riesgo laboral aunque, tal vez, la imagen más popular de este asunto es la transmitida por Lewis Carroll en “Alicia en el País de las Maravillas”, con el personaje del sombrerero loco, término que data del siglo XIX y refleja la constatación de los efectos del envenenamiento habitual de estos artesanos, que usaban soluciones de nitrato de mercurio para ablandar los pelos de los animales, con los que fabricaban los sombreros de fieltro. La revolución industrial y tecnológica de los siglos XIX y XX trajo consigo un gran número de nuevas aplicaciones para el mercurio y muchos de sus compuestos, pero también otras tantas posibles vías de contaminación medioambiental y exposición. La 88 R. P. Mason, W. F. Fitzgerald, F. M. M. Morel, Geochim. Cosmochim. Acta, 1994, 58, 3191-3198. 3. Síntesis de derivados de triazinilglicina. Sensores primera gran señal de alarma se dio con el desastre de la bahía de Minamata (Japón), donde una planta de cloruro de vinilo y acetaldehído estuvo liberando de forma incontrolada grandes cantidades de mercurio en sus aguas residuales desde 1953 a 1960. El resultado fue de un gran número de personas intoxicadas y muertas por la ingestión de pescado contaminado con metilmercurio. Años más tarde, entre 1971 y 1972, más de 400 personas murieron en Irak por intoxicación con metilmercurio. En este caso, el origen se encontraba en el grano que se había usado para hacer el pan, que había sido tratado con un fungicida basado en metilmercurio. Figura 3.3 a y b: Diagramas de flujo representando el ciclo del mercurio en las épocas pre (izquierda) y postindustrial (derecha). Esta serie de desastres sensibilizaron a la comunidad internacional, que se dio cuenta de que la contaminación ejercida por sustancias como las especies de mercurio altamente venenosas puede poner en riesgo nuestro ecosistema, suponiendo una gran amenaza para la salud humana. En particular, las especies organomercúricas (como el metilmercurio) son mucho más virulentas que el Hg (II) inorgánico, debido a que se acumulan rápidamente en varios órganos, igual que cruzan la barrera sangre-cerebro para causar daños en el sistema nervioso central.89 3.2.2. SENSORES90 Muchos cationes y aniones pequeños, que juegan papeles vitales en la vida humana, existen dentro de los organismos y en el ambiente. En consecuencia, la 89 90 M. Santra, B. Roy, K. H. Ahn, Org. Let.., 2011, 13, 13, 3422-3425. J. Du, M. Hu, J. Fan, X. Peng., Chem. Soc. Rev., 2012, 41, 4511-4535. 125 126 3.2. Introducción detección de estos iones es de gran interés e importancia para muchos químicos, biólogos y ambientalistas. Por ejemplo, los iones hierro, sodio y zinc están involucrados funcionalmente en procesos biológicos clave, como la contracción muscular, la transmisión del impulso nervioso o la regulación de la actividad celular.91 Iones como el mercurio o el plomo son tóxicos para los organismos, por lo que su detección pronta es ventajosa. En los últimos años, se han desarrollado numerosas tecnologías analíticas para detectar tales analitos ambientales, médicos y celulares. Por ejemplo, las técnicas basadas en sondas fluorescentes pueden ser tanto sensibles como selectivas. Son también rápidas, fáciles de llevar a cabo en tiempo real, así como económicas. En consecuencia, han recibido una atención especial y se ha realizado un notable progreso. La mayoría de las sondas fluorescentes son sistemas supramoleculares abióticos, que comúnmente se unen a los analitos mediante interacciones no covalentes, como enlaces de hidrógeno, atracciones electrostáticas y coordinación. Dentro de estas sondas fluorescentes, en la literatura podemos encontrar dos tipos de receptores moleculares, llamados quimiosensores y quimiodosímetros, que se han diseñado y usado para estas aplicaciones.92 Los quimiosensores se unen con el analito objetivo a través de interacciones no covalentes (figura 3.4), para dar señales ópticas medibles con una respuesta en tiempo real, normalmente menor de algunos segundos. Estas interacciones suelen ser reversibles cuando los cambios en la concentración del analito están relacionados con los cambios en la cantidad de quimiosensor, tanto libre como ligado. El hecho de que las interacciones sean reversibles presenta ventajas desde el punto de vista de la reutilización. a) B. T.Nguyen, E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118-3127. b) L. A. Cabell, M. D. Best, J. J. Lavigne, S. E. Schneider, D. M. Perreault, M.-K. Monahan, E. V. Anslyn, J. Chem. Soc.,Perkin Trans. 2, 2001, 315-323. c) A. R. Ray, Trends Neurosci., 2006, 29, 200-206. 92 K. Kaur, R. Saini, A. Kumar, V. Luxami, N. Kaur, P. Singh, S. Kumar, Coord. Chem. Rev., 2012, 256, 1992-2028. 91 3. Síntesis de derivados de triazinilglicina. Sensores Figura 3.4. Representación de un quimiosensor. Chae y Czarnik describieron el término quimiodosímetro93 como una molécula abiótica usada para alcanzar el reconocimiento de un analito con la transducción irreversible y concomitante de una señal observable. Esta aproximación implica la reacción de un analito objetivo (anión, catión o molécula neutra) con el quimiodosímetro, y está asociada con una transformación química que implica ruptura y formación de enlaces covalentes. Este proceso resulta en la formación de productos diferentes del quimiodosímetro de partida, que además tienen propiedades ópticas diferentes. Obviamente, sólo en algunos casos se regeneran los quimiodosímetros, usando transformaciones químicas diferentes de las que están implicadas en el proceso de detección analítica. Estas transformaciones químicas, alcanzadas por reacciones específicamente diseñadas, se afectan poco por el entorno, proporcionándonos una clara ventaja en términos de selectividad. Los quimiodosímetros pueden tener dos tipos de mecanismos principales: por un lado, los analitos reaccionan con los quimiodosímetros, mostrando los productos cambios en las señales fluorescentes; por otro lado, los analitos actúan como catalizadores y los quimiodosímetros como sustratos de las reacciones catalizadas. Si el segundo mecanismo puede permitir un mayor cambio, la señal de fluorescencia se amplía mucho más, con menores límites de detección. Tanto los quimiosensores como los quimiodosímetros se pueden clasificar según su mecanismo de actuación en: OFF-ON: cuando la sonda no es fluorescente y la interacción con el analito produce fluorescencia. Figura 3.5a 93 M.Y. Chae, A.W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704-9705. 127 128 3.2. Introducción ON-OFF: cuando la sonda es fluorescente y la interacción con el analito quenchea la fluorescencia. Figura 3.5a. Ilustración esquemática de quimiosensores fluorescentes OFF_ON. Ratiométricos: cuando la sonda es fluorescente y la interacción con el analito modifica las características de fluorescencia, como la longitud de onda. Figura 3.5b Figura 3.5b. Ilustración esquemática de quimiosensores fluorescentes ratiométrico. 3. Síntesis de derivados de triazinilglicina. Sensores 3.3. ANTECEDENTES BIBLIOGRÁFICOS En esta sección únicamente se comentarán algunos de los antecedentes bibliográficos correspondientes a la detección de iones metálicos, debido a que ya se han tratado los antecedentes de síntesis de derivados de 1,3,5-triazina en la sección 2.3 de este trabajo. 3.3.1. DETECCIÓN DE IONES METÁLICOS: Debido a la toxicidad de los iones de metales pesados y de transición, en estos últimos años se ha prestado atención al diseño y la síntesis de receptores artificiales, dirigidos a la detección y reconocimiento de estos tipos de iones metálicos. Gracias a este interés, los sensores químicos fluorescentes se han desarrollado como medio para detectar iones como Hg(II) en muestras biológicas y medioambientales. Se han producido algunos logros exitosos en el desarrollo de sensores químicos fluorescentes de Hg(II), aunque la mayoría de los que se han descrito tienen defectos, particularmente en temas de sensibilidad, selectividad, interferencias con otros iones metálicos o baja solubilidad en disoluciones acuosas.94 Debido a la ingente cantidad de información sobre detección de iones metálicos, repasaremos a continuación sólo algunos artículos representativos sobre el tema, eligiendo preferentemente sensores basados en sistemas de 1,3,5-triazina o que detecten los cationes Hg(II) y Zn(II), materia que será importante más adelante en esta memoria. Hua y colaboradores han sintetizado un quimiosensor “off-on” para Hg(II) basado en un compuesto que contiene 1,3,5-triazina, aunque este heterociclo no tiene nada que ver en la interacción del sensor con Hg(II), como se muestra en la figura 3.6. 94 M.-H. Yang, P. Thirupathi, K.-H. Lee, Org. Let.., 2011, 13, 19, 5028-5031. 129 130 3.3. Antecedentes bibliográficos Figura 3.6. Quimiosensor de Hg(II) con un anillo de 1,3,5-triazina. Se ha visto que la melamina es capaz de interaccionar con iones Hg(II),95 siendo capaz de modificar las propiedades ópticas de una disolución de nanopartículas de oro, dependiendo de la presencia o no de iones Hg(II), como se muestra en la figura 3.7. Figura 3.7. Interacciones entre melamina, Hg(II) y nanopartículas de oro. Misra y colaboradores96 han diseñado una sonda capaz de detectar Hg(II) formando un complejo fluorescente, cuya estructura se puede ver en la figura 3.8. Figura 3.8. Complejo fluorescente de Hg(II). 95 96 J. Du, S. Yin, L. Jiang, B. Ma, X. Chen, Chem. Commun., 2013, 49, 4196-4198. P. Srivastava, R. Ali, S. S. Razi, M. Shahid, S. Patnaik, A. Misra, Tetrahedron Let., 2013, 54, 3688-3693. 3. Síntesis de derivados de triazinilglicina. Sensores Ghosh y colaboradores97 han obtenido un sensor para Cu(II) y Hg(II) basado en rodamina y sulfonamina, cuya estructura se muestra en la figura 3.9. Figura 3.9. Quimiosensor para Cu(II) y Hg(II). Lin y colaboradores98 han diseñado un dendrímero que contiene unidades de 1,3,5-triazina capaz de detectar iones Cu(II). Un esquema representativo del proceso de detección se muestra en la figura 3.10. Figura 3.10. Quimiosensor para Cu(II) basado en triazina. Kumar y colaboradores99 han sintetizado un quimiosensor ratiométrico para Zn(II), cuya estructura mostramos en la figura 3.11, y cuyo mecanismo para la detección es un desplazamiento de la longitud de onda del máximo de emisión. Figura 3.11. Quimiosensor ratiométrico de Zn(II). K. Ghosh, T. Sarkar, A. Samadder, A. R. Khuda-Bukhsh, New. J. Chem, 2012, 36, 2121-2127. M. Sellaiah, Y. C. Rajan, H.-C. Lin, J. Mater. Chem, 2012, 22, 8976-8987. 99 M. Kumar, N. Kumar, V. Bhalla, Chem. Commun., 2013, 49, 877-879. 97 98 131 132 3.3. Antecedentes bibliográficos Han y colaboradores100 han diseñado un quimiosensor ratiométrico de Zn(II), cuyo mecanismo de detección es la formación de agregados entre sistemas aromáticos de pireno. La formación de agregados provoca un desplazamiento batocrómico del máximo de longitud de onda. Meng y colaboradores101 han obtenido un quimiosensor basado en quinoleína capaz de detectar Zn(II) y Cd(II), cuya estructura se muestra en la figura 3.12. N H N N N O N Figura 3.12. Quimiosensor de Zn(II) y Cd(II). 100 101 K. Baek, M. S. Eom, S. Kim, M. S. Han, Tetrahedron Let., 2013, 54, 1654-1657. Y. Cai, X. Meng, S. Wang, M. Zhu, Z. Pan, Q. Guo, Tetrahedron Let.., 2013, 54, 1125-1128. 3. Síntesis de derivados de triazinilglicina. Sensores 3.4. DISCUSIÓN DE RESULTADOS 3.4.1. N,N’-BISARIL-1,3,5-TRIAZINA-2,4,6-TRIAMINAS. 3.4.1.1. Síntesis: Nos planteamos obtener derivados de aminotriazinas con la estructura que se muestra en la figura 3.13, con la intención de usar el grupo amino como nucleófilo en reacciones posteriores. Figura 3.13. N,N’-bisaril-1,3,5-triazina-2,4,6-triaminas. La síntesis de estos compuestos se abordó por dos rutas alternativas que se desarrollaron paralelamente (esquema 3.1). Esquema 3.1. Rutas sintéticas planteadas. 3.4.1.1.1. Ruta 1: Sintetizar derivados amino protegidos con un grupo lábil. 133 134 3.4. Discusión de resultados La introducción de un grupo amino en monoclorotriazinas requiere amoniaco, una temperatura superior a 100ºC, largos periodos de tiempo y material especialmente diseñado para soportar la presión.102 En estas duras condiciones, la radiación microondas se ha mostrado eficiente en la reducción de los tiempos de reacción, el incremento de los rendimientos y evitando la descomposición de reactivos y productos.7 Por ello, en esta primera ruta se plantea obtener derivados de 1,3,5-triazina donde un grupo amino se encuentra protegido por un grupo lábil, cuya desprotección aporta el grupo NH2 libre deseado. Hemos elegido como grupo protector la 2,4-dimetoxibencilamina, porque el grupo bencilo se puede eliminar en una amplia variedad de condiciones (ácido, base y oxidante) para obtener el grupo NH2 libre.103 Las reacciones de 2-cloro-4,6diaminotriazinas con 2,4-dimetoxibencilamina se llevaron a cabo bajo radiación microondas y en ausencia de disolvente. Las triazinas disustituidas de partida se han sintetizado previamente en el grupo de investigación.66 La 2,4-dimetoxibencilamina se usó en un exceso de 2 moles, debido a que ejerce las funciones de nucleófilo y base, para neutralizar el ácido clorhídrico generado en la reacción de sustitución nucleófila aromática. Se optimizaron las reacciones usando tiempos entre 5 y 10 minutos y temperaturas oscilando entre 120 y 150 ºC, utilizando un reactor microondas monomodo. Finalmente, las mejores condiciones se obtuvieron en 5 minutos a 150 ºC (tabla 3.1). Debemos destacar que algunas bencilaminotriazinas se han descrito previamente en condiciones clásicas a 90 ºC en tolueno durante 12 horas.104 En la tabla 3.1 se muestran los mejores resultados obtenidos para cada caso. a) G. Blotny, Tetrahedron, 2006, 62, 9507. b) J. A. Zerkowski, J. P. Mathias, G. M. Whitesides, J. Am. Chem. Soc. 1994, 116, 4305. 7 Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012. 103 P. Kociensky, Protecting Groups, 3rd Ed.; Thieme Verlag: Stuttgart, 2006. 66 A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente, Org. Biomol. Chem. 2003, 1, 4451. 104 E. Hollink, E. E. Simanek, D. E. Bergbreiter, Tetrahedron Let.., 2005, 46, 2005-2008. 102 3. Síntesis de derivados de triazinilglicina. Sensores 135 Tabla 3.1. Síntesis de bencilaminotriazinas. Compuesto Sustituyente Temperatura (ºC) Tiempo (min) Rendimiento (%) 157 5 57 158 5 43 9j 150 5 55 9k 147 5 33 9g 150 10 58 9a NH 9i N 3.4.1.2. N Determinación estructural: Los espectros de 1H-RMN de los productos obtenidos muestran varias señales duplicadas. Este efecto es especialmente importante en los grupos NH y en los protones más próximos a estos grupos, como puede verse a modo de ejemplo en los espectros de los derivados de o- y m-pirazolilfenil (figura 3.14 y figuras en anexo 2). 136 3.4. Discusión de resultados Figura 3.14. Espectros de 1H-RMN a diferentes temperaturas del derivado de m-pirazolilfenil 9i (DMSO). En la figura 3.15 se representan los distintos confórmeros para este tipo de estructuras. Estudios previos105 han mostrado que los confórmeros A y B se observan a baja temperatura, mientras que los C y D no se ven (figura 3.15). 105 A. de la Hoz, A. Sánchez-Migallón, B. T. Pelado, J. R. Ramírez, Arkivoc, en prensa. 3. Síntesis de derivados de triazinilglicina. Sensores Figura 3.15. Posibles confórmeros de los compuestos 9, debido a la rotación restringida del enlace N-triazina. En estos productos, las señales correspondientes a los grupos NH y otras señales duplicadas aparecen con la misma integral. Este hecho nos indica que los confórmeros A y B se intercambian rápidamente a 25 ºC, debido a la menor conjugación del nitrógeno amino bencílico. El incremento de la temperatura produce la coalescencia de todas las señales y, a temperaturas superiores a 80 ºC, sólo se observa un confórmero. Hay que destacar que los protones N-H se desplazan a campo alto (0’5 ppm para los NH-Ar y más de 1 ppm para el NH-Bn), debido probablemente a la menor agregación de los confórmeros a través de enlaces de hidrógeno. Se ha determinado la energía libre de activación en el compuesto 9g, mediante experimentos a temperatura variable (figura 3.16). Estos experimentos se llevaron a cabo incrementando lentamente la temperatura, dando diez minutos para estabilizarla, de cara a determinar la temperatura de coalescencia con un margen de error de 1 ºC. Para el compuesto 9g, la temperatura de coalescencia es de 56 ºC, determinándose la energía libre de activación siguiendo el procedimiento descrito por Sandström,106 obteniendo un valor de 67’18 kJ/mol en este caso. ∆G# = a T [9,972 + log (T/∆ν)] a = 1,914 * 10-2 KJ mol-1 a= 4,575 * 10-3 Kcal mol-1 106 J. Sandström, Dynamic NMR Spectroscopy. Academic Press: New York, 1982, 96. 137 138 3.4. Discusión de resultados 353 K 333 K 313 K ∆G = 67’18 kJ/mol 298 K Figura 3.16. Espectros de 1H-RMN a diferentes temperaturas del derivado de naftaleno 9g (DMSO). Esta energía libre de activación es algo menor que la de otros derivados de 1,3,5triazina relacionados,28 lo que se atribuye a la menor conjugación de estos compuestos, debido a la presencia del grupo bencílico. 28 T. J. Mooibroek, P. Gamez, Inorg. Chim. Acta, 2007, 360, 381. 3. Síntesis de derivados de triazinilglicina. Sensores 3.4.1.3. 139 Ruta 2: Introducción del grupo amino en primer lugar: La sustitución del primer átomo de cloro en el cloruro de cianurilo es una reacción que se produce con facilidad, por ello en esta ruta partimos de cloruro de cianurilo e hidróxido amónico, según describen Chauhan y Giri107 para obtener 10. En una segunda etapa, la aminodiclorotriazina obtenida se hace reaccionar con la amina que nos interese, en una reacción sin disolvente de sólo 10 minutos con microondas. Con este método se ha logrado obtener las triazinas con un grupo amino libre con un rendimiento prácticamente cuantitativo y en unas condiciones medioambientalmente benignas (tabla 3.2). Tabla 3.2. Síntesis de las aminotriazinas 10. R Tiempo (min) Temperatura (ºC) Rto. (%) 10 100 88 10 100 93 30 150 94 Los resultados de esta ruta eran tan buenos que decidimos no llevar a cabo la reacción de desprotección en la ruta 1. Este fue el método elegido para llevar a cabo la síntesis del resto de derivados de aminotriazina. 107 S. M. S. Chauhan, N. G. Giri, Supramol. Chem.20, 8, 2008, 743-752. 140 3.4. Discusión de resultados 3.4.2. N-TRIAZINILGLICINAS 3.4.2.1. Síntesis: Una vez obtenidas las aminotriazinas, llevamos a cabo la reacción con ácido cloroacético, con la idea de sintetizar los derivados de N-triazinilglicina (esquema 3.2): NH2 N R HN + N N 10a-g Cl COOH R N R 11 COOH N N R 13a-g Esquema 3.2. Síntesis de los derivados de N-triazinilglicina. Las propiedades de estos derivados se modularán con una amplia gama de sustituyentes, que incluyen aminas alifáticas y aromáticas mono y disustituidas, así como un compuesto con anillos benzocondensados (figura 3.17). Figura 3.17. Estructura general de los derivados de N-triazinilglicina. Algunas de las condiciones empleadas utilizando la aminotriazina 10c como nucleófilo, para obtener la triazinilglicina 13c se muestran en la tabla 3.3. 3. Síntesis de derivados de triazinilglicina. Sensores 141 Tabla 3.3. Reacción de SN sobre ácido cloroacético para la síntesis de 13c. Entrada Proporción 10c : 11 Temperatura (ºC) Tiempo (min) Disolvente 1 0’5 : 0’6 100 10 No 2 0’5 : 0’6 100 10 DMSO 3 0’5 : 0’6 150 10 DMSO 4 0’5 : 0’6 185 10 DMSO 5 0’5 : 0’6 185 30 DMSO 6ª 0’5 : 0’6 185 30 DMSO a) Con DIPEA En ninguna de las pruebas realizadas hemos observado progreso alguno en la dirección que queremos. Asimismo, se han llevado a cabo pruebas con los derivados de fenilo (10b) y p-nitrofenilo (10h), sin obtención del producto deseado. Estos resultados pueden racionalizarse en base a la baja nucleofilia del grupo amino unido al anillo de triazina, heterociclo π deficiente, que deslocaliza sus electrones en el anillo heteroaromático. Tras ver los resultados negativos anteriores decidimos abordar otra aproximación. En este caso, usaremos como nucleófilo el aminoácido glicina y como sustrato un derivado de cloro-s-triazina, previamente sintetizado en nuestro grupo de investigación (esquema 3.3).66 A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente. Org. Biomol. Chem. 2003, 1, 4451. 66 142 3.4. Discusión de resultados Esquema 3.3. Síntesis de triazinilglicinas. Para poner a punto el método experimental, utilizamos la cloro-s-triazina con sustituyentes de p-anisidina 1c. Los datos se ven reflejados en la tabla 3.4. Tabla 3.4. Síntesis de la triazinilglicina 13c. Entrada Proporción 3:4 Base Temperatura (ºC) Tiempo (min) Disolvente 13c (%) 1 0’5 : 0’5 Na2CO3 185 10 No a 2 0’5 : 0’5 Na2CO3 185 15 No a 3 0’5 : 0’5 DIPEA 185 10 DMSO b 4 0’5 : 0’5 Na2CO3 185 10 DMSO c 5 0’5 : 0’5 Na2CO3 185 15 DMSO c 6 0’5 : 0’5 Na2CO3 185 20 DMSO d 7 0’5 : 0’6 KOH 150 10 DMSO e 8 0’5 : 0’6 KOH 150 3 DMSO 70 a) b) c) d) e) Reacción difícil de controlar, con mezclas complejas de productos. No hay reacción. Reacción incompleta. Producto no aislado. Reacción completa. Producto no aislado (30 % por 1H-RMN). Reacción completa. Mezcla compleja de la que no se ha podido aislar el producto. Las reacciones sin disolvente (entradas 1 y 2, tabla 3.4), en este caso son poco reproducibles y se descontrolan con facilidad. Debido a ello, nos planteamos el uso de DMSO como disolvente (tabla 3.4, entradas 3-8). Por CCF, observamos que la relación 3. Síntesis de derivados de triazinilglicina. Sensores 143 entre reactivos y productos mejora con la fortaleza de la base (entradas 3 vs 4 y entrada 7). La reacción se completa a los 20 minutos (entrada 6, tabla 3.4). No obstante, la CCF y la 1H-RMN nos revelan la existencia de impurezas, que no podíamos eliminar por lavados con disolventes de diferentes características. Una reflexión nos hizo comprender que las impurezas podrían deberse a reacciones colaterales de polimerización. Así, decidimos reducir la temperatura y acotar el tiempo hasta que a los 3 minutos (entrada 8, tabla 3.4) alcanzamos las condiciones óptimas. La purificación del producto ha sido sencilla, ya que precipita puro (70 %) tras añadirle agua acidulada al crudo de reacción (entrada 8). Este método se ha extendido a otros derivados de triazina, obteniendo los rendimientos que pueden verse en la tabla 3.5, junto con las mejores condiciones de reacción para cada caso. Tabla 3.5. Condiciones óptimas para la obtención de las N-triazinilglicinas 13. O HO H N N N R N R R Tiempo (min) Temperatura (ºC) Rendimiento (%) 15 185 80 3 150 70 3 185 70 3 150 70 3 150 60 3 185 70 3 185 70 144 3.4. Discusión de resultados Podemos decir que hemos desarrollado un método medioambientalmente benigno por las siguientes razones: Tiempos cortos de reacción, de 3 minutos en todos los casos, salvo en el del pirazol. Radiación microondas, con lo que implica en un menor consumo de energía. Fácil purificación, mediante un simple lavado con una disolución acuosa. Buen rendimiento, entre el 60 y el 80 % en todos los casos. 3.4.2.2. Determinación estructural: Todos los compuestos fueron caracterizados mediante resonancia magnética nuclear [RMN (1H, 13 C, gHSQC, COSY)], espectroscopia de infrarrojo, puntos de fusión y espectrometría de masas. En los espectros de 1H-RMN se aprecia en todos los casos las señales correspondientes con el grupo amino de la glicina alrededor de 7 ppm, así como la del CH2 de la glicina sobre 4 ppm. También pueden apreciarse las señales correspondientes a los protones de los anillos aromáticos y las del resto de grupos NH en la zona de 9 ppm. En los espectros de 13C se observan señales sobre 165 ppm, típicas del anillo de triazina, y alrededor de 171 ppm, correspondientes al carbono del grupo CO. En lo que respecta a los espectros de infrarrojo, podemos destacar las bandas correspondientes a la vibración de tensión de enlaces N-H en la zona próxima a 3400 cm-1. También podemos apreciar las vibraciones de tensión de los enlaces C=C y C=N entre 1450 y 1600 cm-1, aunque son bandas intensas difíciles de asignar y una banda en torno a 800 cm-1, que corresponde con las vibraciones de deformación del anillo de triazina.82 Además de las mencionadas, se puede observar también una banda sobre 1730 cm-1, correspondiente con el enlace C=O del grupo carboxilo. 82 R. M. Desai, D. K. Dodiya, A. R. Trivedi, V. H. Shah, Med. Chem. Res., 2008, 17, 495-506. 3. Síntesis de derivados de triazinilglicina. Sensores Los espectros de masas muestran picos correspondientes al ión molecular y a las fragmentaciones previsibles para las estructuras de nuestros productos, lo cual representa una prueba importante de que las estructuras que tenemos son las que creemos tener. 3.4.2.3. Estudios de 1H-RMN para el derivado de pirazol: Hemos determinado la energía libre de activación de la sal sódica del derivado de pirazol 13a, mediante experimentos a temperatura variable (figura 3.18), siguiendo el procedimiento descrito por Sandström (ver epígrafe anterior).106 La temperatura de coalescencia de las señales de NH es de 71 ºC. El resultado obtenido para nuestro compuesto ha sido una ∆G‡=71,66 KJ mol-1 y viene a completar los estudios que se han realizado para derivados sustituidos de 2,4-diaminotriazinas57 y ureido-1,3,5- triazinas.68 En la tabla 3.6 se reflejan los desplazamientos químicos a campos más altos de la señal de NH al aumentar la temperatura. Tabla 3.6. Desplazamiento químico de la señal de NH a diferente temperatura. T (°C) 25 35 45 55 65 67 69 70 71 80 NH δ(ppm) 9,35 9,31 9,27 9,24 9,21 9,20 9,19 9,18 9,18 9,15 Figura 3.18. 1H-RMN a diferente temperatura (DMSO, pH 11’1). J. Sandström, Dynamic NMR Spectroscopy. Academic Press: New York, 1982, 96. A. Díaz-Ortiz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, M. C. Mateo, A. Sánchez-Migallón, G. Valiente. New. J. Chem. 2004, 28, 952. 68 A. Ruiz Carretero, J. R. Ramírez, A. Sánchez-Migallón, A. de la Hoz, Eur. J. Org. Chem, submitted manuscript. 106 57 145 146 3.4. Discusión de resultados Cuando el compuesto se aísla por precipitación en medio ácido los espectros de 1 H (figura 3.19) y 13C (figura 3.20) son diferentes a los comentados con anterioridad. Figura 3.19. Espectro de 1H-RMN del compuesto 13a en medio ácido (DMSO, 80ºC). Figura 3.20. Espectro de 13C-RMN del compuesto 13a en medio ácido (DMSO, 80ºC). 3. Síntesis de derivados de triazinilglicina. Sensores Los desplazamientos obtenidos (ver datos experimentales) se entienden suponiendo que la estructura zwitteriónica del aminoácido se formase no por protonación del nitrógeno amínico sino del nitrógeno piridínico del anillo de triazina.108 Por tanto, se podrían formular las estructuras resonantes que se muestran en la figura 3.21 que justificaría el desapantallamiento de todos los NH, así como la dificultad para detectar todos los carbonos del anillo de triazina que sí se observan en la sal sódica de este compuesto. Figura 3.21. Estructuras zwitteriónicas. Un estudio más detallado dependiente del pH se muestra en la figura 3.22. Si tenemos en cuenta que el pKa de la melamina es de 5, el del pirazol es 2’5 y los de la glicina son 2’35 y 9’78, podemos indicar que: - A pHs comprendidos entre 0’9 y 4 se pueden distinguir las señales del grupo carboxilo. Hasta pH 7,4 los espectros son superponibles, salvo por un pequeño apantallamiento del grupo NH de la glicina. - Los cambios más significativos se producen en el intervalo de pHs entre 7,4 y 11,1 donde se observa un claro desplazamiento a campos más altos de las señales que corresponden al grupo NH (de 7,52 ppm a 6,36 ppm) y CH2 (de 3,85 a 3,49) de la glicina. 108 V. Kampyli, D. A. S. Phillips, A. H. M. Renfrew, Dyes Pigments, 2004, 165-175. 147 148 3.4. Discusión de resultados Figura 3.22. Espectros de 1H-RMN de 13a a distintos pHs (DMSO, 25 ºC). - Hay que resaltar que a pH=8,9 todas las señales se ensanchan y podríamos estar cerca del punto isoeléctrico, por eso el espectro a pH 11,1 > pI es el que presenta un δ para el NH sin protonar.109 - Cuando el pH es de 13,1 se puede advertir claramente el apantallamiento de la señal de grupo metileno (3,24 ppm) y un ensanchamiento de todas las señales. 3.4.2.4. Estudios de difracción de Rayos-X: Se han realizado estudios de difracción de Rayos-X para determinar la estructura de los derivados de pirazol 13a y piperidina 13d. El compuesto 13a se ha cristalizado en una mezcla de CH2Cl2 y éter etílico, obteniéndose cristales adecuados para su estudio por difracción de rayos X. En la figura 3.23, se muestra su diagrama ORTEP y en la tabla A3.1. (ver anexo 3) las distancias y ángulos seleccionados. El compuesto cristaliza con una molécula de diclorometano y 109 δ NH2 melamina = 6’2 ppm. 3. Síntesis de derivados de triazinilglicina. Sensores otra de ácido clorhídrico, lo que provoca la protonación del N3 del anillo de triazina, según se había propuesto mediante los estudios de RMN. Los tres grupos NH son coplanares con el anillo de triazina, al igual que el anillo C11-C16, cuyo ángulo diedro con el anillo de triazina es de 3.5º, y se encuentra en una conformación trans con el N2 de la triazina (ángulo de torsión N2-C2-N5-C11 es 178.2(5)º). Sin embargo, el otro anillo aromático (C21-C26) forma un ángulo de 13.1º con el anillo central, y presenta una conformación cis, respecto al átomo N2 de la triazina (ángulo de torsión N2-C3-N8-C21 es 6.8(8)º) (Figura 3.23). Figura 3.23. Diagrama ORTEP para el compuesto 13a con un 30% de probabilidad. Las líneas discontinuas representan los enlaces de hidrógeno. Existe una interesante estructura supramolecular a través de enlaces de hidrógeno y otras interacciones más débiles. Todos los hidrógenos de los nitrógenos, se han localizado en el mapa de densidad electrónica. Como se puede observar en la figura 3.23, hay dos enlaces de hidrógeno intramoleculares entre los grupos NH y los anillos pirazólicos. El compuesto 13a se encuentra formando dímeros a través de enlaces de hidrógeno en los que están implicados tanto el cloruro como el ácido carboxílico (figura 3.24 y tabla 3.7). El Cl1 está implicado en seis enlaces de hidrógeno, dos de ellos 149 150 3.4. Discusión de resultados bifurcados. El primero se produce con N3-H3 y N4-H4 de la misma molécula y el otro enlace bifurcado se produce con C17-H17 y C28-H28, interacciones que tienen lugar con dos moléculas independientes de simetría adecuada. Tabla 3.7. Enlaces de hidrógeno para el compuesto 13a. D-H..A d(D-H) d(H..A) <DHA d(D..A) Simetría O1-H1…Cl1 0.92(6) 2.090 166.5 2.997 1-x, -y, 1-z N3-H3…Cl1 0.61(3) 2.559 151.0 3.110 x, y, z N4-H4…Cl1 0.77(4) 2.543 157.9 2.997 x, y, z C17-H17…Cl1 0.93 2.809 134.6 3.525 1-x, 1-y, 1-z C28-H28…Cl1 0.93 2.810 166.5 3.721 1-x, 1-y, -z C30-H30A…Cl1 0.97 2.574 163.5 3.515 x, y, z C19-H19…O2 0.93 2.619 171.0 3.547 -x, 1-y, 1-z C25-H25…O2 0.93 2.536 156.9 3.411 -x, 1-y, 1-z N5-H5…N7 1.08(3) 1.603 170.3 2.677 x, y, z N8-H8…N10 0.92(4) 1.852 149.5 2.687 x, y, z Figura 3.24. Asociaciones de dímeros, unidos mediante enlaces de hidrógeno. Por otra parte, se observa que el ácido carboxílico se encuentra fuera del plano que define el resto de la molécula (C5 está -1.269 Å fuera del plano de la triazina) y está uniendo mediante interacciones débiles los dímeros, dando lugar a cadenas a lo largo del eje b (figura 3.25). 3. Síntesis de derivados de triazinilglicina. Sensores Figura 3.25. Cadenas formadas a lo largo del eje b. La unión de las cadenas, se ve reforzada por una interacción π−π, face-to-face, entre los anillos aromáticos unidos a la triazina, que se encuentran a 3.776 Å y forman un ángulo diedro de 13.1º (figura 3.26). Figura 3.26. Interacción π−π entre los anillos aromáticos. El compuesto 13d se ha cristalizado en una mezcla de DMSO y MeOH, obteniéndose cristales adecuados para su estudio por difracción de rayos X. En la figura 151 152 3.4. Discusión de resultados 3.27, se muestra su diagrama ORTEP y en la tabla del anexo 3 (A3.2) las distancias y ángulos seleccionados. Figura 3.27. Diagrama ORTEP para el compuesto 13d con un 30% de probabilidad. El protón del ácido carboxílico se ha localizado en el mapa de densidad electrónica protonando el N2 de la triazina, lo que está de acuerdo con la deslocalización de carga observada en el grupo COO, siendo las distancias del enlace de 1.248(4) y 1.231(4) Å para C15-O1 y C15-O2, respectivamente. Hay que destacar, que tanto el grupo amino, como el grupo carboxílico, son coplanares con el anillo de triazina, formando un ángulo diedro de 9.5º entre el anillo de triazina y el plano que forman N6-C14-C15-O1-O2. Existe una interesante estructura supramolecular a través de enlaces de hidrógeno y otras interacciones más débiles. Como se puede observar en la figura 3.28, el compuesto se encuentra formando dímeros a través de enlaces de hidrógeno entre N2H2 y N6-H6, y el oxígeno O1 (figura 3.28 y tabla 3.8). El hidrógeno H13A, forma un enlace bifurcado con ambos oxígenos del grupo carboxílico (O1 y O2). 3. Síntesis de derivados de triazinilglicina. Sensores Tabla 3.8. Enlaces de hidrógeno e interacciones débiles para el compuesto 13d. D-H..A d(D-H) d(H..A) <DHA d(D..A) Simetría N2-H2…O1 0.94 1.751 156.4 2.636 1-x, -y, -z N6-H6…O1 0.86 2.081 144.6 2.823 1-x, -y, -z C13-H13A…O1 0.97 2.542 169.0 3.499 1-x, -y, -z C9-H9A…O2 0.97 2.536 157.5 3.452 x-½, ½+y, z-½ C13-H13A…O2 0.97 2.460 136.1 3.230 1-x, -y, -z Figura 3.28. Asociaciones de dímeros, unidos mediante enlaces de hidrógeno. Por otra parte, se observan interacciones π−π entre los anillos de triazina, con una geometría “offset-face-to-face”, en la que los anillos aromáticos se encuentran desplazados uno respecto a otro. Ello implica una interacción estabilizante entre las nubes π (figura 3.29). La distancia entre los anillos es de 3.519 Å, y forman un ángulo diedro de 0º. 153 154 3.4. Discusión de resultados Figura 3.29. Interacción π−π entre los anillos de triazina. 3.4.2.5. Estudio de las propiedades ópticas: Los resultados obtenidos en los experimentos de UV y fluorescencia de los derivados 13a, 13f y 13g se agrupan en la tabla 3.9. A modo de ejemplo se muestra la gráfica del derivado de pirazol (figura 3.30). 6 UV Fluorescencia 0,4 4 0,2 2 Intensidad (u.a.) Absorbancia 0,3 0,1 0,0 250 300 350 400 450 0 500 Longitud de onda (nm) Figura 3.30. Espectros de UV y fluorescencia de 13a en DCM (10-6 M). 3. Síntesis de derivados de triazinilglicina. Sensores 155 Tabla 3.9. Datos UV y fluorescencia. Absorción Emisión λmáx. (nm) λmáx. [Log ε ] (nm) 13a 242 [5,17] 393 15877 >0,01 13f 259 [4,74] 383 15700 >0,01 13g 234 [4,24] 374 15997 >0,01 Producto R Desplazamiento Stokes (cm-1) ΦF Podemos sacar algunas conclusiones de los espectros anteriores. La primera de ellas es que los máximos de absorción se encuentran en la región del UV, alrededor de 300 nm (tabla 3.9). Este hecho, junto con la alta absortividad molar se atribuye a transiciones π-π*.69 En cuanto a la fluorescencia, el máximo de emisión se localiza por debajo de 400 nm, aunque lo más destacable es que su rendimiento cuántico de fluorescencia es muy bajo, con lo que descartamos que sean aplicables en dispositivos como OLEDs. Se realiza un estudio de solvatocromismo de estos compuestos, probando con disolventes de diferentes características de polaridad. Se realizan espectros UV de las diferentes glicinotriazinas, pudiendo verse una muestra en la figura 3.31. En primer lugar, hay que destacar que estos compuestos son muy poco solubles en disolventes apolares, como el hexano, no alcanzando una concentración suficiente para poder ver un espectro de UV apropiado. En UV apenas hay diferencias en la longitud de onda de los máximos de absorción al variar las características del disolvente. 69 E. Beltrán, J. L. Serrano, T. Sierra, R. Giménez, Org. Let.. 2010, 12, 1404. 156 3.4. Discusión de resultados DCM CH3CN MeOH THF DMSO Absorbance 1,0 0,5 0,0 250 300 350 400 Wavelength (nm) Figura 3.31. Espectros de UV normalizados de 13a con diferentes disolventes. Se realizaron espectros de fluorescencia de las glicinotriazinas en disolventes de distinta polaridad, de los que se muestra un ejemplo en la figura 3.32. En este caso, se ven dos zonas dignas de comentar. La primera de ellas, en torno a 340 nm, contiene máximos de emisión para todos los disolventes, si bien en el caso del DMSO este máximo tiene muy poca intensidad relativa. En la segunda zona, en torno a 390 nm, se observa una alta intensidad de fluorescencia relativa para todos los disolventes polares apróticos, probablemente debido a que estos disolventes pueden favorecer la presencia de agregados. En el caso del MeOH es bastante probable la existencia de enlaces de hidrógeno del propio disolvente con la triazinilglicina. DCM Hex DMSO CH3CN 1,0 MeOH THF Intensity (a.u.) 0,8 0,6 0,4 0,2 0,0 350 400 450 500 550 Wavelength (nm) Figura 3.32. Espectros de fluorescencia normalizados de 13a con diferentes disolventes. 3. Síntesis de derivados de triazinilglicina. Sensores 3.4.2.6. 157 Estudios con iones metálicos: Los sensores y sondas fluorescentes requieren en su estructura química una fracción de reconocimiento y un fluoróforo.72,110 Los compuestos que hemos sintetizado cumplen dichas condiciones, presentando una débil fluorescencia y una alta capacidad para coordinar con metales y, por tanto, pueden ser potenciales quimiosensores. Esto nos ha motivado a estudiar su comportamiento frente a diferentes cationes metálicos. Los resultados obtenidos respecto a una amplia variedad de diferentes cationes metálicos se encuentran recogidos en las figuras 3.33, 3.34 y 3.35. 25 I - I Blanco 20 15 13a 10 5 0 Figura 3.33. Intensidad de fluorescencia de 13a con diferentes cationes. La concentración de los cationes es de 10-5 M, excepto en el caso del Hg (II), que es de 10 -8 M (CH3CN). B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2002 B. Wang and E. J. Anslyn, Chemosensors: Principles, Strategies, and Applications, John Wiley and Sons, New York, 2011. 72 110 158 3.4. Discusión de resultados Figura 3.34. Intensidad de fluorescencia de 13f con diferentes cationes. La concentración de los cationes es de 10-5 M, excepto en el caso del Hg (II), que es de 10 -8 M (CH3CN). Figura 3.35. Intensidad de fluorescencia de 13g con diferentes cationes. La concentración de los cationes es de 10-5 M, excepto en el caso del Hg (II), que es de 10 -8 M (CH3CN). Como muestra la figura 3.33 el derivado de pirazol 13a, que posee un nitrógeno en el anillo π-excedente y que, por tanto, presenta una mayor capacidad de 3. Síntesis de derivados de triazinilglicina. Sensores 159 coordinación, es el que interacciona con mayor variedad de cationes metálicos. En este mismo sentido, el compuesto 13f manifiesta afinidad por cationes Hg(II), Zn(II) y Cu(II) entre otros (figura 3.34.). Sin embargo, el derivado 13g revela una alta afinidad por el catión Hg(II), por ello, se va a realizar un estudio más detallado para su aplicación como quimiosensor de dicho catión (figura 3.35). 3.4.2.6.1. Sonda de mercurio: El comportamiento del derivado de naftaleno 13g muestra una respuesta fluorescente selectiva y altamente sensible hacia el catión Hg(II), y por eso, se va a investigar, entre otros, el comportamiento de dicho compuesto frente al pH, el límite de detección de Hg(II), la estequimetría entre el ligando y el catión, también se va a buscar la concentración de agregación crítica (CAC). Los espectros de UV y fluorescencia del derivado de naftiltriazinilglicina 13g se recogen en la figura 3.36. 0,35 60 UV Fluorescencia 0,30 50 0,25 40 Abs. 30 0,15 20 0,10 Intensidad (u.a.) 0,20 10 0,05 0,00 300 400 0 500 Longitud de onda (nm) Figura 3.36. Espectros UV y fluorescencia de 13g (CH3CN, 10-5 M). En ausencia de mercurio, la sonda (10-7M) presenta un máximo de emisión a 380 nm cuando se irradia a 278 nm. En presencia de concentraciones superiores a 10-9 M de Hg(II) la intensidad de fluorescencia aumenta considerablemente (aproximadamente un 160 3.4. Discusión de resultados orden de magnitud), y aparece un nuevo máximo a 334 nm junto al de 380 nm, lo que indica una respuesta ratiométrica, como se refleja en el esquema 3.4. Esquema 3.4. Derivado de naftaleno 13g sin y con Hg(II) (CH3CN). Como el nuevo máximo se encuentra desplazado hacia la zona UV del espectro electromagnético, se observa un cambio de color en la disolución de la sonda con Hg(II) que refleja esta desplazamiento hipsocrómico (fotografía del esquema 3.4). La compatibilidad medioambiental es un factor primordial a la hora de encontrar aplicación en biosistemas. Por ello, es importante destacar que esta sonda es bastante soluble en medios acuosos a pH fisológico (PBS: CH3CN 9:1),111 condición necesaria para su uso en la detección de mercurio en agua y organismos vivos. A continuación, estudiamos cómo influye la concentración de Hg(II) en la fluorescencia. A una concentración de la sonda de 10-7 M en PBS: CH3CN 9:1, se le adicionan disoluciones de HgCl2 en PBS en un rango de concentraciones comprendido entre 10-10 y 2x10-7 M. Los resultados obtenidos se muestran en la figura 3.37, revelando un incremento de la intensidad de fluorescencia en el máximo a 380 nm conforme aumenta la concentración de Hg(II), además de la aparición de un nuevo máximo a 334 nm para concentraciones Hg(II) superiores a 2x10-9 M. Con los datos de los espectros de concentración variable de HgCl2 se ha representado una curva (figura 3.38), que muestra un cambio pronunciado en la señal de fluorescencia cuando la concentración de Hg(II) está alrededor de 2x10-9 M, es decir, 111 PBS (Tampón fosfato salino, pH= 7,4). 3. Síntesis de derivados de triazinilglicina. Sensores 161 tenemos una sonda para Hg(II) a nivel nanomolar. En la misma figura mostramos una ampliación del rango lineal de la valoración, para poder calcular el límite de detección del método (LD). Los valores a partir de los que se ha representado la gráfica se muestran en la tabla A3.3, en el anexo 3. Blanco -9 10 M -9 2x10 M -9 3x10 M -7 10 M 120 Intensity (a.u.) 100 80 60 40 20 0 300 350 400 450 Wavelength (nm) Figura 3.37. Espectros de fluorescencia de 13g con diferentes concentraciones de Hg(II) (PBS:CH3CN 9:1). 120 Intensidad (a.u.) 100 80 -10,5 -9,5 -8,5 Log [Hg (II)] -7,5 -6,5 60 150 40 100 20 50 0 0 1.000E-12 y = 7E+10x - 70,176 R² = 0,9892 2.000E-12 [Hg (II)] Figura 3.38. a) Representación de la intensidad de fluorescencia de una disolución 10-7 M de 13g en PBS:CH3CN 9:1, con diferentes concentraciones de Hg (II). b) Ampliación del rango lineal. El límite de detección (LD) se define como el nivel de concentración de analito más bajo que proporciona en el instrumento una señal estadísticamente diferente a la 162 3.4. Discusión de resultados señal de un blanco analítico112 (ver parte experimental). La buena correlación lineal (R2 = 0,9892) entre el valor de la intensidad de fluorescencia y la concentración de Hg(II) en el rango de 1’2 a 2‘2 nM resulta en un límite de detección lineal de 1’2 nM, por debajo del límite que marca la EPA para contaminación en agua potable (10 nM) y agua industrial (250 nM).113 Para validar la elevada selectividad de la sonda hacia el ion mercúrico se llevaron a cabo experimentos competitivos adicionando ion Hg 2+ a disoluciones de la sonda 13g en presencia de iones Ag(I), Ba(II), Ca(II), Cd(II), Cs(I), Cu(I), Cu(II), Fe(II), Fe(III), Mn(IV), Sn(II), Zn(II), Al(III), Na(I), K(I) y Mg(II). Los resultados obtenidos se muestran en la figura 3.3- y muestran que dichos metales no interfieren en la emisión de fluorescencia del complejo Hg 2+ - Ligando. Éste es un logro muy importante porque con frecuencia los quimiosensores de Hg(II) presentan interferencias con Ag(I) o Cd(II). 114 a) IUPAC. Compendium of Chemical Terminology. 2nd Ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications. Oxford. 1997. b) International Union of pure and Applied Chemistry. Nomenclature, Symbols, Units and Their Usage in Spectrochemical Analysis. 2. Data Interpretation. Spectrochimica Acta. 33, 242 (1978). c) J.N Miller and J. C. Miller, Estadística y Químiometria para la Química Analítica Prentice Hall, Madrid, 2002. 113 Mercury Update: Impact of Fish Advisories. EPA Fact Sheet EPA-823-F-01-011; EPA, Office of Water: Washington, DC, 2001. 114 Algunas revisiones recientes: a) D. T. Quang, J. S. Kim, Chem. Rev., 2010, 110, 6280; b) X. Chen, X. Tian, I. Shin, J. Yoon, Chem. Soc. Rev., 2011, 40, 4783; c) A. Razgulin, N. Ma, J. Rao, Chem. Soc. Rev., 2011, 40, 4186; d) Z. Liu, W. He, Z. Guo, Chem. Soc. Rev., 2013, 42, 1568-1600. 112 3. Síntesis de derivados de triazinilglicina. Sensores 163 Hg (II) + otros cationes 100 90 80 I - I blanco 70 60 Catión 50 Catión + Hg (II) 40 30 20 10 Hg(II) Mg(II) K(I) Na(I) Al(III) Zn(II) Sn(II) Mn(IV) Fe(III) Fe(II) Cu(II) Cu(I) Cs(I) Cd(II) Ca(II) Ba(II) Ag(I) 0 Figura 3.36. Respuesta fluorescente de 13g (10-6 M) a la adición de Hg(II) (10-8 M) o de otros iones metálicos (10-5 M) (barras azules) y a la mezcla de otros iones metálicos (10-5 M) con Hg(II) (10 -8 M) (barras moradas) en CH3CN. Existen varios métodos para calcular la estequiometria del complejo formado entre la sonda y el catión. De estos métodos usaremos el de Job,115 que consiste en mezclar diferentes cantidades de las disoluciones de sonda y catión, con la única restricción de que el volumen total de la mezcla debe ser constante, es decir, la suma de las concentraciones de sonda y catión debe ser constante. Los resultados se muestran en la figura 3.39. Según la representación de Job, la estequiometria del complejo formado sería de, aproximadamente, 4 moléculas de sonda por cada una de HgCl2, lo que podría indicar que la sonda forma agregados y en su interior se alojarían los cationes Hg2+. 115 P. Job, Ann. Chim.-Rome Appl., 1928, 9, 113-203. 164 3.4. Discusión de resultados Método Job Hg(II) 0,16 0,14 Abs. 280 nm 0,12 0,1 0,08 0,06 0,04 0,02 0 0 0,2 0,4 0,6 0,8 1 X Hg(II) Figura 3.39. Representación de Job de la absorbancia de la sonda frente a la fracción molar de Hg2+ (PBS:CH3CN 9:1). También hemos estudiado la sonda a diferentes valores de pH, encontrando que este sensor es útil para detectar Hg(II) en un amplio intervalo de pH (2-12), ya que su intensidad de fluorescencia permanece prácticamente constante en este rango. Como en el capítulo 2, se ha estudiado la posibilidad de que los derivados de triazinilglicina formen agregados. Para ello, se han realizado los estudios con Rojo Nilo y de DLS, técnicas cuyo fundamento está explicado en la sección 2.3.1.3.5 de esta memoria. En el experimento con Rojo Nilo se ha obtenido la gráfica que se muestra en la figura 3.40. En este caso, la CAC es de 1’9x10-5 M. 3. Síntesis de derivados de triazinilglicina. Sensores 165 60 Intensidad (u.a.) 50 40 y = -1,708x + 35,68 R² = 0,3283 30 y = -9,3787x - 0,5583 R² = 0,8745 20 10 0 -7 -6 -5 -4 -3 -2 Log [13g] Figura 3.40. Representación del estudio de 13g con Rojo Nilo (CH3CN). Se realizó un estudio de DLS del compuesto 13g, preparando una disolución de concentración 10-4 M en acetonitrilo (figura 3.41). Este experimento demuestra que existen agregaciones con una población mayoritaria que tiene un diámetro hidrodinámico de 217 nm, dato que concuerda con la experiencia del Rojo Nilo, ya que se ha hecho este estudio de DLS a una concentración superior a la que antes obtuvimos como CAC. Figura 3.41. Distribución de diámetro hidrodinámico de 13g (CH3CN, 10-4 M). 166 3.4. Discusión de resultados Se preparó una disolución de concentración 10-7 M de 13g para realizar la medida de DLS pero, como se esperaba, no aparecen agregados, ya que la CAC es del orden de 10-5 M. Ensayos en células:116 3.4.2.6.2. Viendo la eficacia de nuestra sonda en la detección de mercurio, y el amplio rango de pH en el que puede actuar, se decidió probar a introducirla en un sistema vivo para ver si sigue manteniendo sus propiedades. El sistema vivo elegido es un cultivo de células endoteliales humanas de cordón umbilical. En el ensayo realizado con la sonda 13g no se observa ningún cambio entre las medidas realizadas sin sonda y las realizadas con sonda, lo que parece indicarnos que ésta no es capaz de atravesar la membrana celular. En este punto, se decidió realizar una modificación química de la sonda, transformando el grupo ácido carboxílico, muy polar, en un grupo éster, menos polar, que con mayor probabilidad permitiría a la sonda atravesar la membrana celular. En el interior de la célula, las enzimas de tipo esterasa hidrolizarían la sonda esterificada, recuperando la sonda original, tal y como sucede para muchas sondas comerciales como Fluo-4, Fura-2, MitoSOX, etc (www.lifetechnologies.com). Se planificó la síntesis del producto esterificado como se indica en el esquema 3.5: NH2 O HN Cl N Cl O HN O N N O H2N N Cl Cl N N O N 8g HN N N Cl 15 14g Esquema 3.5. Síntesis de la sonda esterificada 14g. 116 Ensayos realizados con la colaboración del Dr. Mario Durán, profesor de la Facultad de Medicina. O NH 3. Síntesis de derivados de triazinilglicina. Sensores El derivado 15 se obtuvo con un rendimiento del 60 %. Sin embargo, la segunda etapa fue infructuosa, porque se obtenían mezclas de productos difíciles de separar en todas las condiciones de reacción empleadas para llevar a cabo la sustitución nucleófila de los átomos de cloro de la 4,6-diclorotriazin-2-ilglicina (15) por naftilamina (8g). Por ello, se modificó la estrategia sintética a la que se muestra en el esquema 3.6. Esquema 3.6. Síntesis de la sonda esterificada 14g. El derivado 1g, se obtuvo siguiendo el método de síntesis de monoclorotriazinas propuesto por Kolmakov.117 Llegar al derivado esterificado 14g fue más complicado, porque en las diferentes condiciones de reacción empleadas para sustituir el tercer átomo de cloro de la triazina 1g se producía la hidrólisis del grupo éster. Después de bastantes pruebas experimentales donde se combinaron distintos tiempos, temperaturas, bases y proporción de reactivos se llegó a una solución de compromiso entre hidrólisis del éster y la finalización de la reacción. Los datos se recogen en la tabla 3.10: El empleo de bases débiles, como DIPEA o carbonato sódico da lugar a conversiones más pobres y concuerda con los resultados encontrados en la literatura.118 La base fuerte ayuda a desplazar el equilibrio de protonación del aminoácido esterificado y lo hace más nucleófilo. Las condiciones óptimas de reacción resultaron ser 3 minutos a 150 ºC, empleando un equivalente KOH y una relación 1g/16 de 5:6. Aunque la reacción no se había completado, en estas condiciones se consiguió evitar la hidrólisis del éster. 117 118 K. A. Kolmakov, J. Heterocyclic Chem., 2008, 45, 533-539. W. Karuehanon, W. Fanfuenha, A. Rujiwatra, M. Pattarawarapan, Tetrahedron Let.., 2012, 53, 3486-3489. 167 168 3.4. Discusión de resultados Tabla 3.10. Resumen de las experiencias para la síntesis de 14g. Entrada Proporción Base 1g:16 Temperatura Tiempo (ºC) (min) Disolvente 14g (%) 1 1 : 1’2 KOH 140 10 No 2 1 : 2’4 DIPEA 185 15 DMSO 3 1 : 2’4 DIPEA 185 2 DMSO 4 1 : 1’2 Na2CO3 185 15 DMSO 5 1 : 1’2 Na2CO3 185 20 DMSO 6 1 : 1’2 KOH 185 3 DMSO 7 1 : 1’2 KOH 150 10 DMSO 8 1 : 1’2 KOH 150 3 DMSO 40 La purificación del producto no era evidente. En primer lugar, se adicionó HCl 0’1 M y se centrifugó el crudo. A partir de aquí se realizaron pruebas con numerosos disolventes para aislar el producto, resultando ser diclorometano y éter etílico los disolventes más adecuados. Tras este proceso se obtenía el producto puro con un 40 % de rendimiento. Una vez obtenido el producto esterificado 14g, se realizaron ensayos con un cultivo de células endoteliales. Para la realización del ensayo, se siguieron las pautas descritas en la parte experimental. Por limitaciones técnicas, debido a la zona de emisión de nuestra sonda, y a la ausencia de láseres que permitan la excitación de la muestra en ese rango, no se pudo obtener imágenes con un microscopio confocal, por ello se han representado los espectros de fluorescencia adquiridos en las figuras 3.42 y 3.43. 3. Síntesis de derivados de triazinilglicina. Sensores 169 60 Intensidad (u.a.) Células control -6 Células + sonda 10 M 40 20 0 350 400 450 500 Longitud de onda (nm) Figura 3.42. Emisión de las células control y con sonda 10-6 M en PBS:DMSO (1000:1). 800 Células (control) -6 Células con Hg 10 M -6 Células, sonda y Hg (ambos 10 M) Intensity (a.u.) 600 400 200 0 350 400 450 500 Wavelength (nm) Figura 3.43. Emisión de fluorescencia de células, sonda (10-6 M) y Hg (II) (10-6 M) en PBS:DMSO (1000:1). Las células endoteliales se han incubado durante una hora en medio de cutivo SmBM conteniendo 10-6 M de 14g en PBS: DMSO (1000:1). Se elige este tiempo para alcanzar una solución de compromiso entre concentración de la sonda en el interior de las células y la muerte de las mismas, debido a la toxicidad de la sonda y disolventes. En este mismo sentido, se ha elegido DMSO en vez CH3CN para disminuir la toxicidad. Tras esta incubación, se reemplazó el medio de cultivo por medio fresco durante media hora para permitir la hidólisis del enlace éster por parte de las esterasas celulares. 170 3.4. Discusión de resultados En la figura 3.42 se observa una pequeña diferencia entre la emisión de fluorescencia que presentan las células endoteliales control y en presencia de nuestra sonda. Esta pequeña diferencia puede indicar que la sonda ha penetrado en el interior de la célula, aunque no podemos determinar en qué proporción. Por otro lado, se sabe que el ión Hg2+ penetra en las células más rápidamente que la sonda porque lo hace a través de canales iónicos a favor de gradiente. El resultado obtenido cuando las células se incuban solo con ión Hg2+ muestra un gran incremento de la intensidad de fluorescencia (figura 3.43), hecho que se podría justificar por una posible interacción del Hg(II) con dinucleótidos como NADH, NADPH o aminoácidos aromáticos y proteínas119 y cromatina.120 Cuando se introduce nuestra sonda en el cultivo endotelial y se añade el ion Hg2+ se aprecian dos hechos: 1) Una pequeña disminución de la intensidad de fluorescencia respecto a las células con Hg(II) sin sonda. Esta tendencia aumenta con la concentración de la sonda figura 3.44. Una posible explicación sería una mayor afinidad del ión Hg(II) por nuestra sonda frente a las moléculas orgánicas mencionadas anteriormente. Sin sonda -6 Sonda 10 M -6 Sonda 2x10 M 800 Intensidad (u.a.) 600 400 200 0 350 400 450 Longitud de onda (nm) Figura 3.44. Emisión de las células con Hg(II) 10-6 M variando la concentración de sonda, en PBS:DMSO (1000:1). 119 120 R. F. Chen, Arch. Biochem. Biophys., 1971, 142, 2, 552–564. S. E. Bryan, A. L. Guy, K. J. Hardy, Biochemistry-US, 1974, 13, 2, 313–319. 3. Síntesis de derivados de triazinilglicina. Sensores 171 2) La aparición de una nueva banda de emisión a una longitud de onda mayor (347 nm), lo que demuestra que la sonda 14g interacciona con Hg2+ dentro de la célula. 3.4.2.6.3. Sonda de zinc: En primer lugar, en la figura 3.45 se muestran los espectros de UV y fluorescencia del producto 13b. 8 UV Fluorescencia 0,15 7 6 Absorbancia 4 3 0,05 Intensidad (u.a.) 5 0,10 2 1 0,00 300 400 0 500 Longitud de onda (nm) Figura 3.45. Espectros de UV y fluorescencia de 13b (CH2Cl2, 10-5 M). El Zn2+ es el segundo metal de transición más abundante en el cuerpo humano y, como se ha comentado anteriormente, juega un papel vital en numerosos procesos biológicos, lo que da importancia al descubrimiento de nuevos quimiosensores para la detección de dicho catión. En el diseño de sondas fluorescentes, el reto más importante es discriminar diferentes iones metálicos con propiedades químicas similares, como sucede entre el Zn2+ y el Cd2+. Estos metales presentan, básicamente, la misma química de coordinación y es muy difícil distinguirlos.121 Es importante destacar que el derivado de fenilo 13b se comporta como una sonda que diferencia completamente estos iones, presentando una alta selectividad a) Z.-K. Song, B. Dong, G.-J. Lei, M.-J. Peng, Y. Guo, Tetrahedron Lett., 2013, 54, 4045-4949 b) K. Tsukamoto, S. Iwasaki, M. Isaji, H. Maeda, Tetrahedron Lett., 54, 5971-5973. 121 172 3.4. Discusión de resultados hacia el Zn (II). Además de revelarse como una sonda específica para Zn (II) -figura 3.46-, nuestra sonda muestra una respuesta ratiométrica para este catión, como se puede apreciar en el esquema 3.7. 25 I- I blanco 20 15 10 5 Ag(I) Al(III) Ba(II) Ca(II) Cd(II) Cs(I) Cu(I) Cu(II) Fe(II) Fe(III) Hg(II) K(I) Mg(II) Mn(IV) Na(I) Sn(II) Zn(II) 0 Figura 3.46. Respuesta fluorescente de 13b (10-6 M) con diferentes cationes metálicos (10-5 M) en (CH3CN). Esquema 3.7. Respuesta ratiométrica del derivado de N-feniltriazinilglicina 13b en presencia de Zn2+ en CH3CN. Este sensor, en presencia de Zn(II), ve incrementada su intensidad de fluorescencia, como se puede apreciar de forma sutil en la fotografía (figura 3.47). 3. Síntesis de derivados de triazinilglicina. Sensores 173 Figura 3.47. Fluorescencia de 13b en presencia (izquierda) y en ausencia (derecha) de Zn(II) (CH3CN). De la misma forma que con la sonda de Hg(II), con el Zn(II) se ha realizado un experimento variando la concentración del catión, manteniendo la concentración de sonda en 10-7 M. En el rango de la medida estudiado, apenas se observan diferencias en los espectros de fluorescencia registrados (figura 3.48), por lo que no se ha podido calcular el límite de detección de esta sonda. Blanco -9 Zn 10 M -8 Zn 10 M -8 Zn 5x10 M -7 Zn 10 M -7 Zn 2x10 M -6 Zn 10 M -5 Zn 10 M -4 Zn 10 M 35 34 33 32 Intensity (a.u.) 31 30 29 28 27 26 25 24 23 22 21 20 300 350 400 450 Wavelength (nm) Figura 3.48. Fluorescencia del compuesto 13b con diferentes concentraciones de Zn(II) (PBS:CH3CN 9:1). Para calcular la estequiometria, se realizaron medidas aplicando el método de Job (figura 3.49), anteriormente descrito. El punto en el que se alcance la absorbancia máxima es el que indica la estequiometria del complejo. 174 3.4. Discusión de resultados Método Job Zn(II) 0,35 y = 0,4468x + 0,0725 R² = 0,9136 0,3 Abs. 0,25 y = -0,2657x + 0,4067 R² = 0,9158 0,2 0,15 0,1 0,05 0 0 0,2 0,4 0,6 0,8 1 XZn(II) Figura 3.49. Representación de Job de la absorbancia de la sonda frente a la fracción molar de Zn2+ (PBS:CH3CN 9:1). Según la gráfica de Job, el complejo debería tener una estequiometria 1:1 de ligando y de catión. Para tratar de averiguar qué tipo de complejo se obtiene en este caso, se hizo interaccionar el complejo con ZnCl2 para obtener un sólido cuya estructura debería corresponder al complejo. A este sólido le se intentó determinar su estructura mediante espectroscopia de 1H-RMN (figuras 3.50 y 3.51) o IR (figura 3.52). Comparando los espectros de 1H-RMN (figura 3.50), no se observan cambios significativos en el desplazamiento de las señales, sólo podemos apreciar un sutil apantallamiento de las señales correspondientes a los grupos NH en presencia de Zn(II). Comparando los espectros de 13 C-RMN (figura 3.51) se pueden observar cambios apreciables en las señales correspondientes a los carbonos del anillo de triazina, así como al carbono carboxílico, manteniéndose el resto de señales inalteradas, lo que parece indicar una coordinación del catión Zn (II) con los nitrógenos del anillo de triazina y el/los oxígenos del grupo carboxilo. 3. Síntesis de derivados de triazinilglicina. Sensores Figura 3.50. 1H-RMN de 13b con Zn(II) –abajo- y sin Zn(II) –arriba- (DMSO). Figura 3.51. 13C-RMN de 13b con Zn(II) –abajo- y sin Zn(II) –arriba- (DMSO). 175 176 3.4. Discusión de resultados 100 Sólido CH3CN11 JR-151 %T 98 96 94 92 90 88 Ligando 86 Ligando + Zn2+ 84 82 80 3600 JR15 3300 3000 2700 2400 2100 1950 1800 1650 1500 1350 1200 1050 900 750 600 1/cm Figura 3.52. Espectros IR de 13b con (negro) y sin (azul) Zn(II). En los espectros de IR también se observan algunos cambios en presencia de Zn(II). La intensidad de las bandas entre 1000 y 1350 cm-1 aumenta en presencia de Zn(II), mientras en la región del espectro entre 1400 y 1650 cm-1 las bandas son más intensas en ausencia de Zn(II). Otro cambio significativo se da en la zona de 3300 cm-1. En ausencia de Zn(II) observamos una banda estrecha y definida mientras que, en presencia de Zn(II), la banda es mucho más ancha. En resumen, vemos cambios en las bandas del anillo de triazina y en las de NH. Con esta información, proponemos una estructura con un entorno tetraédrico para el átomo de Zn como la que se muestra en la figura 3.53 para el complejo de 13b y Zn(II). 3. Síntesis de derivados de triazinilglicina. Sensores 177 Figura 3.53. Estructura propuesta para el complejo de 13b y Zn(II). 3.4.3. REACCIONES CON NANOESTRUCTURAS DE CARBONO: Tal como se planteaba en los objetivos de este capítulo, y basándonos en la experiencia de nuestro grupo de investigación, se planteó la funcionalización del buckminsterfullereno (o fullereno de C60), usando para ello una reacción de cicloadición 1,3-dipolar (esquema 3.8), con los aminoácidos sintetizados en este trabajo. Esquema 3.8. Reacción de cicloadición 1,3-dipolar sobre C60. Para poner a punto el método experimental, se utilizó la glicinotriazina 13d, con la que se realizaron pruebas que se recogen en la tabla 3.11. 178 3.4. Discusión de resultados Las condiciones iniciales de reacción (entrada 1, tabla 3.11) consistieron en usar tolueno a reflujo (110 ºC) durante 12 horas. Tras este tiempo, no se observa cambio alguno en el matraz de reacción, ya que la disolución morada con la que se inicia la reacción permanece morada. Observando que con tolueno la reacción no experimentaba progresos, se decide cambiar a un disolvente similar que nos permita alcanzar una temperatura mayor. En las entradas 2 y 3 de la tabla 3.11 se elige 1,2-diclorobenceno como disolvente, que permite llegar a 180 ºC. Cuando la reacción alcanza las 18 horas, se observa por CCF la aparición de varias manchas diferentes del C60, por lo que se decide cortar la reacción para evitar en lo posible polisustituciones. Tabla 3.11. Resumen de las experiencias más representativas de la reacción de 13d y C60. Entrada Proporción C60 : 13d : A1 Disolvente Temperatura Tiempo Rto 15d (%) 1 1 : 1’2 : 6 Tolueno 110 ºC 12 h No reacciona 2 1 : 1’2 : 6 1,2-diclorobenceno 180 ºC 12 h No reacciona 3 1 : 3 : 12 1,2-diclorobenceno 180 ºC 18 h 12 % Una vez finalizada la reacción, para aislar el producto se realiza una columna en cromatografía, usando tolueno como eluyente e incrementando la polaridad progresivamente con mezclas de tolueno y acetato de etilo. Aunque la columna elimina algunas impurezas, aún sigue habiendo una mezcla de productos, que se intenta separar con ciclos de lavado con CH2Cl2 o éter etílico y centrifugación. Tras este proceso, se 3. Síntesis de derivados de triazinilglicina. Sensores obtiene el producto deseado puro, con un 12 % de rendimiento, aunque no se pudo secar por métodos convencionales, debido a la gran afinidad del C60 por los disolventes utilizados. Lavando el producto con CDCl3, para desplazar los disolventes del C60, solamente se consigue introducir nuevas impurezas. Se extendió la reacción al derivado de naftaleno 13g, en las mismas condiciones con las que hemos obtenido los mejores resultados para la reacción con el derivado de piperidina. En este caso, el rendimiento fue del 11 %. En resumen, aunque se ha obtenido el producto que buscábamos, no se ha podido obtener con la pureza suficiente para poder realizar pruebas posteriores. Se han registrado los espectros de 1H-RMN de estos derivados, observando la señal correspondiente a los CH2 del anillo de pirrolidina a un desplazamiento de 5’6 ppm, lo que es indicativo de que se ha obtenido el producto deseado. 179 180 3.5. Parte experimental 3.5. PARTE EXPERIMENTAL 3.5.1. EQUIPAMIENTO. En el apartado 2.5.1 se describen los diferentes equipos utilizados. 3.5.2. SÍNTESIS DE MONOTRIAZINAS CON 2,4- DIMETOXIBENCILAMINA N-(2,4-Dimetoxibencil)-N´,N´´-bis-(2-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (9a). En un matraz para el microondas provisto de refrigerante de reflujo se introduce 6-cloro-2,4-bis-(2-pirazol-1-ilfenilamino)-1,3,5-triazina (0’11 g, 0’25 mmol) y 2,4dimetoxibencilamina (0’084 g , 0’50 mmol). La mezcla se irradia durante 5 minutos a 50 W alcanzándose una temperatura máxima de 157 ºC. El crudo de reacción se enfría y se lava con 3 ml de etanol. El sólido resultante se filtra a vacío. Se obtienen 0’08 g (57%) de N-(2,4-dimetoxibencil)-N´,N´´-bis-(2-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6triamina. 1 H-RMN (DMSO, ppm) T=95 ºC δ: 3’75 (s, 3H,- OCH3); 3’81 (s, 3H, -OCH3); 4’37 (d, J=5’37 Hz, 2H, N-CH2); 6’44 (d, J=7’32 Hz, 1H, H5’Ph.); 6’53 (s, 2H, H4 pir.); 6’56 (s, 1H, H3’Ph.); 7’10 (d, J=7’8 Hz, 1H, H6’Ph.); 7’17 (t, J=7’56 Hz, 3H, H4 Ph. y NH); 7’32 (s ancho, 2H, H5 Ph.); 7’48 (d, J=7’8Hz, 2H, H3 Ph.); 7’84 (s, 2H, H3 pir.); 8’13 (s, 2H, H5pir.); 8’29 (s ancho, 2H, H6 Ph.); 9’10 (s ancho, 2H, NH). 13 C-RMN (DMSO, ppm) T=95 ºC. δ= 38’12 (N-CH2) ; 54’85 (-OCH3) ; 55’17 (OCH3); 98’31 (C3’Ph.); 104’44 (C5’Ph.); 106’43 (C4 pir.); 119’37 (C1’Ph.); 122’58 (C6 Ph.); 123’27 (C4 Ph.); 123’38 (C3 Ph.); 126’82 (C5 Ph.); 128’42 (C6’Ph.); 129’84 (C2 Ph.); 130’66 (C5 pir.); 132’05 (C1 Ph.); 140’31 (C3 pir.); 157’51 (C4’Ph.); 159’35 (C2’Ph.); 163’74 (C2 Tz); 165’58 (C4,6 Tz). MS (MALDI-TOF): m/z (%) = 561’293 (100) [M+H]+. Punto de fusión: 114-117 ºC. IR (Neto) υ (cm-1): 3421’72 (NH); 1506’41 y 1408’04 (C=N and C=C); 1205 (OCH3). 3. Síntesis de derivados de triazinilglicina. Sensores 181 N2-(2,4-Dimetoxibencil)-N4,N6-di(naftalen-1-il)-1,3,5-triazina-2,4,6-triamina (9g). En un matraz para el microondas provisto de refrigerante de reflujo se introduce 6-cloro-2,4-bis-(naftilamino)-1,3,5-triazina (0’99 g, 0’25 mmol) y 2,4- dimetoxibencilamina (0’084g , 0’50 mmol). La mezcla se irradia durante 10 minutos a 150ºC. El crudo de reacción se enfría y se lava con 2x3 ml de HCl (0.1M). El sólido resultante se filtra a vacío. La purificación se lleva a cabo mediante cromatografía en columna de gel de sílice utilizando como eluyente hexano:acetato (9:1) gradiente acetato. Se obtienen 0’077 g (58%) de N2-(2,4-dimetoxibencil)-N4,N6-di(naftalen-1-il)1,3,5-triazina-2,4,6-triamina. 1 H-RMN (DMSO, ppm) T=80 ºC. δ: 3’73 (d, J=2’2 Hz, 3H, OCH3); 3’75 (d, J=2’2 Hz, 3H, OCH3); 4’29 (s ancho, 2H, N-CH2); 6’36 (s, 1H, H5’ Ph); 6’50 (s, 1H, H3’ Ph); 6’65 (s, 1H, NH); 6’94 (s, 1H, H6’ Ph); 7’36 (d, J=5’1 Hz, 2H, H2 Naph.): 7’48 (d, J=3’6 Hz, 4H, H6,7 Naph.); 7’63 (s, 2H, H4 Naph.) ; 7’67 (d, J=6’5 Hz, 4H, H3 Naph); 7’89 (s, 2H, H5 Naph); 8’04 (s, 2H, H8 Naph); 8’63 (s, 2H, NH). 13 C-RMN (DMSO, ppm) T=80ºC. δ: 38’12 (N-CH2); 54’90 (OCH3); 55’09 (OCH3); 98’13 (C3 Ph); 104’18 (C5 Ph); 119’78 (C1 Ph); 122’00 (C4 Naph) 122’78 (C8 Naph) 123’95 (C3 Naph); 124’98 (C6,7 Naph); 125’22 (C2 Naph); 127’46 (C5 Naph); 128’52 (C6 Ph); 157’48 (C4 Ph); 159’26 (C2 Ph); 165’62 (C4,6 Tz); 166’03 (C2 Tz). MS (MALDI-TOF): m/z (%) = 529’177 (100) [M+H]+. Punto de fusión: 83-86 ºC. IR (Neto) υ (cm-1): 3426 (NH); 1587; 1479 (C=N y C=C); 1263 (OCH3); 1034 (OCH3). N-(2,4-Dimetoxibencil)-N´,N´´-bis-(3-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (9i). En un matraz para el microondas provisto de refrigerante de reflujo se introduce 6-cloro-2,4-bis-(3-pirazol-1-ilfenilamino)-1,3,5-triazina (0’11g, 0’25mmol) y 2,4dimetoxibencilamina (0’084g , 0’50mmol). La mezcla se irradia durante 5 minutos a 50 W alcanzándose una temperatura máxima de 157 ºC. El crudo de reacción se enfría y se le añaden 4 ml de agua, se introduce en un baño de ultrasonidos durante 15 minutos y 182 3.5. Parte experimental el sólido resultante se filtra a vacío. La purificación se lleva a cabo mediante cromatografía en columna de gel de sílice utilizando como eluyente hexano:acetato (1:1). Se obtienen 0’06g (43%) de N-(2,4-dimetoxibencil)-N´,N´´-bis-(3-pirazol-1ilfenil)-1,3,5-triazina-2,4,6-triamina. 1 H-RMN (DMSO, ppm) T=100 ºC. δ: 3’76 (s, 3H, -OCH3); 3’82 (s, 3H, -OCH3); 4’55 (s, 2H, N-CH2); 6’46-6’48 (m, 2H, H3’ y H5’Ph.); 6’58 (d, J=2’44 Hz, 2H, H4 pir.); 6’97 (s ancho, 1H, NH); 7’20 (d, J=8’29Hz, 1H, H6’ Ph); 7’31(t, J=8’05Hz, 2H, H5 Ph.); 7’38 (d, J=7’81Hz, 2H, H4 Ph.); 7’68(d, J=1’46Hz, 2H, H3 pir.); 7’73(d, J=7’81Hz, 2H, H6 Ph.);8’17(s, 2H, H5 pìrazol); 8’20( s, 2H, H2 Ph.); 8’99 (s ancho, 2H, NH). 13 C-RMN (DMSO, ppm) T=100 ºC. δ: 38’33 (N-CH2); 54’88 (OCH3); 55’14 (OCH3); 98’39 (C3’Ph.); 104’53 (C5’Ph.); 106’92 (C4 pir.); 110’36 (C2 Ph.);111’93 (C4 Ph.); 117’72 (C6 Ph.); 119’44 (C1’Ph.); 126’96 (C5 Ph.); 128’26 (C6’Ph.); 128’68 (C5 pir.); 139’65 (C3 Ph.); 140’08 (C3 pir.); 140’68 (C1 Ph.); 157’54 (C4’Ph.); 159’45 (C2’Ph.); 163’75 (C2 Tz); 165’41 (C4,6 Tz). MS (MALDI-TOF): m/z (%) = 561’426 (100) [M+H]+. Punto de fusión: 115-118 ºC. IR (Neto) υ (cm-1): 3446 (NH); 1583; 1506 (C=N y C=C); 1022 (OCH3). N-(2,4-Dimetoxibencil)-N´,N´´-bis-(4-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (9j). En un matraz para el microondas provisto de refrigerante de reflujo se introduce 6-cloro-2,4-bis-(4-pirazol-1-ilfenilamino)-1,3,5-triazina (0’08 g, 0’19 mmol) y 2,4dimetoxibencilamina (0’063 g , 0’38 mmol). La mezcla se irradia durante 5 minutos a 50 W, alcanzándose una temperatura máxima de 150 ºC. El crudo de reacción se enfría y se lava con 3 ml de etanol, disolviéndose el producto. La purificación se lleva a cabo mediante cromatografía en columna de gel de sílice utilizando como eluyente hexano:acetato (1:1). Se obtienen 0’057 g (55%) de N-(2,4-dimetoxibencil)-N´,N´´-bis(4-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina. 3. Síntesis de derivados de triazinilglicina. Sensores 1 H-RMN (DMSO, ppm) T=80 ºC. δ: 3’72 (s, 3H, OCH3); 3’80 (s, 3H, OCH3); 4’46 (d, J=5’8 Hz, 2H, N-CH2); 6’46 (dd, J=8’3 y J=2’4 Hz 1H, H5’Ph); 6’48 (t, J=2’4 Hz, 2H, H4 pir.); 6’55 (d, J=2’5 Hz, 1H, H3’ Ph); 6’93 (t, J=5’8 Hz, 1H, NH); 7’15 (d, J=8’3Hz, 1H, H6’ Ph); 7’62 (d, J=8’8 Hz, 4H, H2 y 6 Ph.): 7’66 (d, J=1’4 Hz, 2H, H3 pir.); 7’80 (d, J=8’3 Hz, 4H, H3 y 5 Ph.) ; 8’23 (d, J=2’5 Hz, 2H, H5 pir.); 8’88 (s ancho, 2H, NH). 13 C-RMN (DMSO, ppm) T=80 ºC. δ: 38’81 (N-CH2); 55’38 (OCH3); 55’65 (OCH3); 98’64 (C3’Ph.); 104’87 (C5’Ph.); 107’44 (C4 pir.); 119’04 (C2,6 Ph.); 119’92 (C1’Ph.); 120’92 (C3,5 Ph.); 127’44 (C5 pir.); 128’62 (C6’Ph.); 134’44 (C4 Ph.); 138’51 (C1 Ph.); 140’52 (C3 pir.); 157’96 (C4’Ph.); 159’43 (C2’Ph.); 164’27 (C4,6 Tz); 166’08 (C2 Tz). MS (MALDI-TOF): m/z (%) = 561’243 (100) [M+H]+. Punto de fusión: 90-93 ºC. IR (Neto) υ (cm-1): 3415 (NH); 1520; 1504 (C=N y C=C); 1207 (OCH3); 1032 (OCH3). N-(2,4-Dimetoxibencil)-N´,N´´-bis-(1-fenilpirazol-4-il)-1,3,5-triazina-2,4,6-triamina (9k). En un matraz para el microondas provisto de refrigerante de reflujo se introduce 6-cloro-2,4-bis-(1-fenilpirazol-4-ilamino)-1,3,5-triazina (0’11g, 0’25mmol) y 2,4dimetoxibencilamina (0’084g , 0’50mmol). La mezcla se irradia durante 5 minutos a 50W alcanzándose una temperatura máxima de 145ºC. El crudo de reacción se enfría y se lava con 3 ml de etanol y el sólido insoluble se filtra a vacío. Se obtienen 0’058 g (40%) de N-(2,4-dimetoxibencil)-N´,N´´-bis–(1-fenilpirazol-4-il)-1,3,5-triazina-2,4,6triamina. 1 H-RMN (DMSO, ppm) T=115 ºC. δ: 3’76 (s, 3H, OCH3); 3’83 (s, 3H, OCH3); 4’55 (d, J=6’34 Hz, 2H, N-CH2); 6’50 (d, J=8’29 Hz, 1H, H5’Ph.); 6’60 (s, 1H, H3’Ph.); 6’97 (s ancho, 1H, NH); 7’21 (d, J=8’78 Hz, 1H, 183 184 3.5. Parte experimental H6’Ph.); 7’26 (t, J=7’32 Hz, 2H, H4 Ph.); 7’45 (t, J=7’32Hz, 4H, H3 Ph.); 7’70 (s ancho, 4H, H2 Ph.); 7’83 (s,2H, H3 pir.); 8’51 (s ancho, 2H, H5 pir.); 8’80 (s ancho, 2H, NH). 13 C-RMN (DMSO, ppm) T=115 ºC. δ: 38’06 (N-CH2); 54’83 (OCH3); 55’13 (OCH3); 98’42 (C3’Ph.); 104’60 (C5’Ph.); 116’79 (C5 pir.); 117’41 (C2,6 Ph.); 119’63 (C1’Ph.); 124’72 (C6’Ph.); 124’95 (C4 Ph.); 127’94 (C4 pir.); 128’71 (C3,5 Ph.); 133’33 (C3 pir.); 139’65 (C1 Ph.); 157’32 (C4’Ph.); 159’32 (C2’Ph.); 163’45 (C4,6 Tz); 166’12 (C2 Tz). MS (MALDI-TOF): m/z (%) = 561.284 (100) [M+H]+. Punto de fusión: 197-200ºC. IR (Neto) υ (cm-1): 3427 (NH); 1558; 1506 (C=N y C=C); 1211 (OCH3); 1032 (OCH3). 3.5.3. SÍNTESIS DE 6-AMINOTRIAZINAS-2,4- DISUSTITUIDAS En un matraz de microondas se pesan 0’5 mmol de 4,6-dicloro-1,3,5-triazina-2amina y 2’5 mmol de la anilina correspondiente para cada caso y se irradia con microondas, en las condiciones indicadas en cada caso. Tras esta etapa, se adicionan al crudo de reacción unos 5 ml de agua y se introduce en un baño de ultrasonidos durante 15 minutos. Una vez disgregado el sólido, se filtra a vacío, se lava con agua (2x5 ml) y se seca a vacío. 4,6-Dicloro-1,3,5-triazina-2-amina (10):109 A partir de cloruro de cianurilo (2,4,6-tricloro-1,3,5-triazina) y una disolución de hidróxido amónico 1 M. En un matraz de fondo redondo de 100 ml se introducen 2’5 g (13’5 mmol) de cloruro de cianurilo, adicionando unos 20 ml de acetona. Acto seguido se adicionó lentamente, gota a gota, 30 ml de una disolución de hidróxido amónico 1M, manteniendo la temperatura de la mezcla de reacción por debajo de 5 ºC con un baño de hielo. Una vez terminada la adición, se deja agitar durante 30 minutos más, momento en 109 S. M. S. Chauhan, N. G. Giri, Supramol. Chem., 2008, 20, 743-752. 3. Síntesis de derivados de triazinilglicina. Sensores el que se retira el baño de hielo y se deja estar la reacción otros 30 minutos a temperatura ambiente. El sólido obtenido se filtra a vacío, se lava con agua (4 x 25 ml) y se seca a vacío para obtener un sólido blanco (1’56 g, 70 %). 1 H-RMN (DMSO, 25 ºC) δ: 8’55 (s, 2H, NH2). Punto de fusión: 224-227 ºC. IR (Neto) υ (cm-1): 796, 1012, 1504, 1643, 3219, 3304, 3387. N2,N4-Difenil-1,3,5-triazina-2,4,6-triamina (10b):122 A partir de 4,6-dicloro-1,3,5-triazina-2-amina (0’082 g, 0’5 mmol) y anilina (0’233 g, 2’5 mmol). Se introduce el matraz en el microondas durante 10 minutos a 100 ºC. El producto se purificó siguiendo el procedimiento descrito, para obtener un 88 % de rendimiento (0’122 g). 1 H-RMN (DMSO, 25 ºC) δ: 6’56 (s, 2H, NH2), 6’95 (t, 2H, J = 7’3 Hz, H4Ph), 7’24 (t, 4H, J = 7’7 Hz, H3,5Ph), 7’80 (d, 4H, J = 7’7 Hz, H2,6Ph), 9’03 (s, 2H, NH). 13 C-RMN (DMSO, 25 ºC) δ: 119’89 (C2,6 Ph), 121’51 (C4 Ph), 128’25 (C3,5 Ph), 140’30 (C1 Ph), 164’43 (C2,4 Tz), 166’81 (C6 Tz). Punto de fusión: 182-184 ºC, descompone. IR (Neto) υ (cm-1): 1217, 1228, 1363, 1737, 2968. N2,N4-Bis(4-metoxifenil)-1,3,5-triazina-2,4,6-triamina (10c):123 A partir de 4,6-dicloro-1,3,5-triazina-2-amina (0’082 g, 0’5 mmol) y p-anisidina (0’307 g, 2’5 mmol). Se introduce el matraz en el microondas durante 10 minutos a 100 ºC. El producto se purificó siguiendo el procedimiento general, para obtener un 93 % de rendimiento (0’157 g). 1 H-RMN (DMSO, 25 ºC) δ: 3’71 (s, 3H, OCH3), 6’39 (s ancho, 2H, NH2), 6’82 (d, 4H, J = 8’8 Hz, H3,5 Ph), 7’62 (d ancho, 4H, J = 7’4 Hz, H2,6 J. A. Zerkowski, J. C. MacDonald, C. T. Seto, D. A. Wierda, G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 2382. 123 F. H. S. Curd, J. K. Landquist, F. L. Rose, J. Chem. Soc., 1947, 154. 122 185 186 3.5. Parte experimental Ph), 8’78 (s, 2H, NH). 13 C-RMN (DMSO, 25 ºC) δ: 55’13 (OCH3), 113’45 (C3,5 Ph), 121’66 (C2,6 Ph), 133’40 (C1 Ph), 154’28 (C4 Ph), 164’40 (C2,4 Tz), 166’79 (C6 Tz). Punto de fusión: 176-179 ºC, descompone. IR (Neto) υ (cm-1): 818, 1217, 1228, 1366, 1666, 1737, 2968. N2,N4-Bis(4-nitrofenil)-1,3,5-triazina-2,4,6-triamina (10h): A partir de 4,6-dicloro-1,3,5-triazina-2-amina (0’082 g, 0’5 mmol) y pnitroanilina (0’345 g, 2’5 mmol). Se introduce el matraz en el microondas durante 30 minutos a 150 ºC. El producto se purificó siguiendo el procedimiento general, para obtener un 94 % de rendimiento (0’173 g). 1 H-RMN (DMSO, 25 ºC) δ: 7’22 (s ancho, 2H, NH2), 8’09 (d ancho, 4H, J = 8’7 Hz, H2,6 Ph), 8’18 (d, 4H, J = 8’7 Hz, H3,5 Ph), 10’12 (s ancho, 2H, NH). 13 C-RMN (DMSO, 25 ºC) δ: 119’13 (C2,6 Ph), 124’64 (C3,5 Ph), 141’01 (C4 Ph), 146’58 (C1 Ph), 163’61 (C2,4 Tz), 165’84 (C6 Tz). Punto de fusión: 173-176 ºC, descompone. IR (Neto) υ (cm-1): 1217, 1229, 1364, 1737, 2968. 3.5.4. SÍNTESIS GENERAL DE DERIVADOS DE NTRIAZINILGLICINA En un matraz de microondas se pesan 0’5 mmol de la triazina monoclorada correspondiente en cada caso, junto con 0’6 mmol (0’045 g) de glicina y 0’060 g de KOH al 85 %. Se adicionan 0’5 ml de DMSO. Se irradia microondas en las condiciones indicadas en cada caso, acoplando al matraz un refrigerante de reflujo. Tras la reacción, se adicionan unos 5 ml de HCl 0’1 M al matraz, formándose un sólido, que se disgrega con la ayuda de un baño de ultrasonidos y se filtra a vacío. En cada caso se indica si es necesaria una etapa posterior de purificación y el rendimiento obtenido en las reacciones. 3. Síntesis de derivados de triazinilglicina. Sensores Ácido 2-((4,6-bis((2-(1H-pirazol-1-il)fenil)amino)-1,3,5-triazin-2-il)amino)acético (13a): A partir de 6-cloro-N,N'-bis-(2-pirazol-1-ilfenil)-[1,3,5]-triazina-2,4-diamina (0’5 mmol, 0’220 g). Se introduce el matraz en el microondas durante 15 minutos a 185 ºC. Para purificar el producto, se disuelve el crudo en diclorometano y se añade una disolución acuosa de NaOH hasta poner pH básico, precipitando el producto deseado (13a), que se obtiene por filtración a vacío (0'188 g, 80 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 25 ºC) δ: 3’50 (d, J = 4’4 Hz, 2H, CH2), 6’38 (s, 1H, NH gly), 6’56 (s, 2H, H4 pir.), 7’18 (m, J = 4’4 Hz, 2H, H4 Ph), 7’37 (m, J = 4’88 Hz, 2H, H5 Ph), 7’50 (d, J = 7’8 Hz, 2H, H3 Ph), 7’89 (d, J = 7’3 Hz, 2H, H3 pir.), 8’22 (s, 2H, H5 pir.), 8’34 (m, 2H, H6 Ph), 9’30 (s, 1H, NH), 9’39 (s, 1H, NH). 13 C-RMN (DMSO, 25 ºC) δ: 45’21 (CH2), 107’14 (C4 pir.), 123’08 (C4 Ph), 123’76 (C6 y C3 Ph), 127’45 (C5 Ph), 129’90 (C2 Ph), 131’33 (C5 pir.), 132’18 (C1 Ph), 141’00 (C3 pir.), 163’71 (C Tz), 164’03 (C Tz), 164’68 (C Tz), 171’11 (CO). MS (FAB): m/z 469’1 [M+H]+; HRMS calculado para C23H21N10O2 m/z: 469’1849, encontrado 469’1852. Punto de fusión: 178-179 ºC. IR (Neto) υ (cm-1): 1454, 1494, 1506, 1714, 3309. Ácido 2-(4,6-bis(fenilamino)-1,3,5-triazin-2-il)amino)acético (13b): A partir de 6-cloro-N,N'-difenil-[1,3,5]-triazina-2,4-diamina (0’5 mmol, 0'148 g). Se introduce el matraz en el microondas durante 3 minutos a 185 ºC, obteniéndose el producto deseado (13b) siguiendo los pasos descritos en el procedimiento general (0'118 g, 70 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’99 (s, 2H, CH2), 6’97 (s, 2H, H4 Ph), 7’11 (s, 1H, NH Gly), 7’26 (s, 4H, H3,5 Ph), 7’74 (s, 4H, H2,6 Ph), 8’98 (s, 2H, NH Ph). 187 188 3.5. Parte experimental 13 C-RMN (DMSO, 80 ºC) δ: 42’0 (CH2), 120’3 (C2,6 Ph), 122’2 (C4 Ph), 128’0 (C3,5 Ph), 139’2 (C1 Ph), 164’1 (C4,6 Tz), 171’1 (COOH). MS (FAB): m/z 337’0 [M+H]+; HRMS calculado para C17H16N6O2 m/z: 337’1413, encontrado 337’1416. Punto de fusión: 223-225 ºC, descompone. IR (Neto) υ (cm-1): 752, 1444, 1494, 3292. - Complejo de 13b con zinc: A partir de 13b se ha sintetizado el complejo con Zn(II). Partiendo de ácido 2(4,6-bis(fenilamino)-1,3,5-triazin-2-il)amino)acético (1 mmol, 0’337 g) y de cloruro de zinc (1 mmol, 0’136 g). En un matraz de 100 ml se mezclan los reactivos, adicionando 30 ml de acetonitrilo como disolvente. La mezcla se calienta a reflujo (80 ºC) durante una hora. Tras esta etapa, se elimina el disolvente en el rotavapor y se seca el producto a vacío. Se obtienen 0’473 g de un sólido blanco. 1 H-RMN (DMSO, 80 ºC) δ: 3’99 (s, 2H, CH2), 6’97 (s, 2H, H4 Ph), 7’11 (s, 1H, NH Gly), 7’26 (s, 4H, H3,5 Ph), 7’74 (s, 4H, H2,6 Ph), 8’98 (s, 2H, NH Ph). 13 C-RMN (DMSO, 80 ºC) δ: 42’0 (CH2), 120’3 (C2,6 Ph), 122’2 (C4 Ph), 128’0 (C3,5 Ph), 139’2 (C1 Ph), 164’1 (C4,6 Tz), 171’1 (COOH). MS (MALDI-TOF): m/z 337’0 [M+H]+. Punto de fusión: 99-103 ºC. IR (Neto) υ (cm-1): 1026, 1224, 1450, 1631, 1734, 3421. Ácido 2-((4,6-bis((4-metoxifenil)amino)-1,3,5-triazin-2-il)amino)acético (13c): A partir de 6-cloro-N,N'-bis-(4-metoxifenil)-[1,3,5]-triazina-2,4-diamina (0’5 mmol, 0'178 g). Se introduce el matraz en el microondas durante 3 minutos a 150 ºC, obteniéndose el producto deseado (13c) siguiendo los pasos descritos en el procedimiento general (0'139 g, 70 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 25 ºC) δ: 3’73 (s, 6H, OCH3); 3’92 (s, 2H, CH2); 6’83 (m, 4H, H3,5 Ph); 7’61 (s ancho, 4H, H2,6Ph); 8’94 (s, 1H, NH); 12’51 (s ancho, 1H, COOH). 3. Síntesis de derivados de triazinilglicina. Sensores 13 C-RMN (DMSO, 25 ºC) δ: 42’1 (CH2); 55’2 (OCH3); 113’5 (C3,5Ph); 121’8 (C2,6Ph); 133’1 (C1Ph); 154’5 (C4Ph); 163’8 (C4,6Tz); 165’49 (C2Tz); 172’13 (COOH). MS (FAB): m/z 397’3 [M+H]+; HRMS calculado para C19H20N6O4 m/z: 397’1624, encontrado 397’1628. Punto de fusión: 246-250 ºC, descompone. IR (Neto) υ (cm-1): 1031, 1238, 1487, 1508, 3294. Ácido 2-((4,6-di(piperidin-1-il)-1,3,5-triazin-2-il-amino)acético (13d): A partir de 6-cloro-N,N'-bis-piperidino-[1,3,5]-triazina-2,4-diamina (0’5 mmol, 0’140 g). Se introduce el matraz en el microondas durante 3 minutos a 150 ºC, obteniéndose el producto deseado (13d) siguiendo los pasos descritos en el procedimiento general (0'112 g, 70 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 25 ºC) δ: 1’43 (s ancho, 8H, H3,5 Piperidina); 1’56 (s, 4H, H4 Piperidina); 3’60 (s ancho, 8H, H2,6 Piperidina); 3’78 (d, 2H, J = 5’86 Hz, CH2); 6’85 (t, 1H, J = 6’10 Hz, NH); 12’32 (s ancho, 1H, COOH). 13 C-RMN (DMSO, 25 ºC) δ: 24’5 (C4 Piperidina); 25’4 (C3,5 Piperidina); 42’3 (CH2); 43’4 (C2,6 Piperidina); 164’4 (C4,6 Tz); 165’9 (C2 Tz); 172’4 (COOH). MS (FAB): m/z 321’1 [M+H]+; HRMS calculado para m/z: 321’2039, encontrado 321’2032. Punto de fusión: 198-202 ºC. IR (Neto) υ (cm-1): 1284, 1597, 1674, 2937, 3267. Ácido 2-((4,6-dimorfolino-1,3,5-triazin-2-il)amino)acético (13e): A partir de 6-cloro-N,N'-bis-morfolino-[1,3,5]-triazina-2,4-diamina (0’5 mmol, 0'142 g). Se introduce el matraz en el microondas durante 3 minutos a 150 ºC, obteniéndose el producto deseado (13e) siguiendo los pasos descritos en el procedimiento general (0'098 g, 60 %). 189 190 3.5. Parte experimental Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’57 (s, 8H, H3,5 Morfolina), 3’61 (s, 8H, H2,6 Morfolina), 3’82 (d, J = 5’85 Hz, 2H, CH2 Gly), 7’01 (t, J = 6’07 Hz, 1H, NH) 13 C-RMN (DMSO, 80 ºC) δ: 42’3 (CH2 Gly), 43’2 (C2,6 Morfolina), 66’0 (C3,5 Morfolina), 164’6 (C4,6 Tz), 165’7 (C2 Tz), 172’1 (COOH). MS (FAB): m/z 325’1 [M+H]+; HRMS calculado para C13H20N6O4 m/z: 325’1624, encontrado 325’1615. Punto de fusión: 203-204 ºC. IR (Neto) υ (cm-1): 1114, 1556, 1672, 3296. Ácido 2-(4,6-bis(difenilamino)-1,3,5-triazin-2-il)amino)acético (13f): A partir de 6-cloro-N2,N2,N4,N4-tetrafenil-1,3,5-triazina-2,4-diamina (0’5 mmol, 0’224 g). Se introduce el matraz en el microondas durante 3 minutos a 185 ºC, obteniéndose el producto deseado (13f) siguiendo los pasos descritos en el procedimiento general (0'171 g, 70 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’61 (s, 2H, CH2), 6’89 (s, 1H, NH), 7’17 (s, 4H, H4 Ph), 7’23 (s, 8H, H2,6 Ph), 7’28 (s, 8H H3,5 Ph). 13 C-RMN (DMSO, 80 ºC) δ: 41’4 (CH2), 124’6 (C4 Ph), 127’3 (C2,6Ph), 128’0 (C3,5 Ph), 143’3 (C1 Ph), 165’3 (C2 Tz), 170’9 (COOH). MS (ESI): m/z 489’2 [M+H]+; HRMS calculado para C29H25N6O2 m/z: 489’2033, encontrado 489’2028. Punto de fusión: 237-241 ºC. IR (Neto) υ (cm-1): 690, 1392, 1537, 1548, 3417. Ácido 2-((4,6-bis(naftalen-1ilamino)-1,3,5-triazin-2-il)amino)acético (13g): A partir de 6-cloro-N2,N4-di(naftalen-1-il)-1,3,5-triazina-2,4-diamina (0’5 mmol, 0’198 g). Se introduce el matraz en el microondas durante 3 minutos a 185 ºC, 3. Síntesis de derivados de triazinilglicina. Sensores 191 obteniéndose el producto deseado (13g) siguiendo los pasos descritos en el procedimiento general y, finalmente, lavando tres veces con fracciones de 2 ml de acetona (0'153 g, 70 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3.89 (d, J = 4.4, 2H, CH2), 6.92 (s ancho, 1H, NH gly), 7.37 (t, J = 7.81 Hz, 2H, H2), 7.49 (m, 4H, H6 y H7), 7.64 (d, J = 8.3 Hz, 2H, H4), 7.68 (d, J = 6.89 Hz, 2H, H3), 7.88 (d, J = 7.56 Hz, 2H, H5), 8.03 (d, J = 6.3, 2H, H8), 8.89 (s, 2H, NH). 13 C-RMN (DMSO, 80 ºC) δ: 47.72 (CH2), 122.14 (C4), 122.70 (C8), 124.25 (C3), 125.02 (C2), 125.15 (C6), 125.31 (C7), 127.52 (C5), 128.47 (C8a), 133.47 (C4a), 134.20 (C1), 164.8 (C4 y C6 Tz), 165.23 (C2 Tz), 171.14 (CO). MS (FAB): m/z 437’1 [M+H]+; HRMS calculado para C25H20N6O2 m/z: 437’1726, encontrado 437’1724. Punto de fusión: 234-236, cambio de fase. IR (Neto) υ (cm-1): 1392, 1556, 1699, 3348, 3392. 2-((4,6-bis(naftalen-1ilamino)-1,3,5-triazin-2-il)amino)acetato de metilo (14g): A partir de 6-cloro-N2,N4-di(naftalen-1-il)-1,3,5-triazina-2,4-diamina (0’25 mmol, 0’099 g) y glicinato de metilo (0’3 mmol, 0’027 g). A la mezcla en el matraz de microondas le añadimos KOH (0’25 mmol, 0’014 g). Se introduce el matraz en el microondas durante 3 minutos a 150 ºC, En primer lugar, adicionamos HCl 0’1 M y centrifugamos el crudo. El sólido resultante se lava con diclorometano (2 ml) y éter etílico (2 ml). Tras este proceso se obtenía el producto deseado (14g) puro con un 40 % de rendimiento (0'045 g, 40 %). Datos físicos y espectroscópicos: 1 H-RMN (DMSO, 80 ºC) δ: 3’44 (s, 3H, CH3), 3’75 (s, 2H, CH2), 7’04 (s ancho, 1H, NH gly), 7’27 (s, 192 3.5. Parte experimental 2H, H2), 7’38 (s ancho, 4H, H6 y H7), 7’44 (s ancho, 2H, H4), 7’57 (s ancho, 2H, H3), 7’78 (s ancho, 2H, H5), 7’89 (s ancho, 2H, H8), 8’83 (s, 1H, NH), 8’91 (s, 1H, NH). 13 C-RMN (DMSO, 80 ºC) δ: 41’81 (CH2), 51’50 (CH3), 122’52 (C4), 123’40 (C8), 124’40 (C3), 125’37 (C2), 125’54 (C6), 125’69 (C7), 127’87 (C5), 128’76 (C8a), 134’72 (C4a), 134’90 (C1), 165’59 (C4 Tz), 165’99 (C6 Tz), 166’16 (C2 Tz), 171’13 (CO). MS (FAB): m/z 450’2 [M+H]+; HRMS calculado para C26H23N6O2 m/z: 451’1877, encontrado 451’1883. Punto de fusión: 101-104 ºC. IR (Neto) υ (cm-1): 1215, 1394, 1556, 1747. 3.5.5. EXPERIMENTOS DE UV Y FLUORESCENCIA MEDIDAS A pH VARIABLE: En un vaso de precipitados de 800 ml se preparan 400 ml de una disolución 10-7 M de la sonda, usando como disolvente una mezcla de acetonitrilo y agua en proporción 1:9. A esta mezcla se adicionan pequeñas cantidades de HCl concentrado y de lentejas de NaOH para ir variando el pH de la disolución modificando el volumen lo menos posible, para mantener la concentración de la sonda prácticamente constante. De esta disolución se toman alícuotas de 2 ml para efectuar las medidas de UV y fluorescencia. VALORACIONES: Se prepara una disolución madre de la sonda 10-3 M y otra del catión metálico (Hg2+ o Zn2+ en nuestro caso) de la misma concentración, usando en ambos casos acetonitrilo como disolvente. Se diluye la sonda hasta tener una disolución de concentración 10-6 M, de la que se toman 0,2 ml para introducirlos en la cubeta de fluorescencia. Se adicionan 1,8 ml de PBS (tampón fosfato salino, de pH fisiológico) para tener en la cubeta una disolución de concentración 10-7 M de la sonda, con una 3. Síntesis de derivados de triazinilglicina. Sensores proporción de disolventes acetonitrilo:agua de 1:9. Se miden las propiedades ópticas de esta disolución, tomándolas como blanco. Al blanco se adicionan cantidades crecientes del catión metálico en estudio (Hg2+ o Zn2+). Para ello, se añaden volúmenes reducidos de disoluciones más concentradas, para que el volumen total de la disolución no experimente cambios apreciables. Por ejemplo, si en la cubeta debe haber una concentración de Hg2+ de 10-7 M, se adicionan 2 µl de la disolución de concentración 10-4 M de este catión. REPRESENTACIÓN DE JOB: Se preparan disoluciones de sonda y de catión de concentración 10-6 M en ambos casos, usando como disolvente una mezcla de acetonitrilo y agua en proporción 1:9. Se mezclan en la cubeta de fluorescencia diferentes cantidades de ambas disoluciones, con la única restricción de que el volumen total de la mezcla debe ser de 2 ml, es decir, la suma de las concentraciones de sonda y catión debe ser constante. LÍMITE DE DETECCIÓN El límite de detección (LD) se define como el nivel de concentración de analito más bajo que proporciona en el instrumento una señal estadísticamente diferente a la señal de un blanco analítico. Por ello, para calcular el límite de detección, es imprescindible definir en primer lugar qué señal es “estadísticamente diferente a la del blanco”. El criterio más utilizado para métodos de calibración univariante es el recomendado por la IUPAC (“International Union of Pure and Applied Chemistry”) en 1978,112b [1] según el cual el límite de detección es aquella concentración de analito que proporciona una señal neta igual a tres veces la desviación estándar del blanco, tal y como indica la ecuación LD = 112b 3s B b International Union of pure and Applied Chemistry. Nomenclature, Symbols, Units and Their Usage in Spectrochemical Analysis. 2. Data Interpretation. Spectrochimica Acta. 33, 242, 1978. 193 194 3.5. Parte experimental donde sB es la desviación estándar del blanco, b la pendiente de la recta de calibrado y 3 un factor de seguridad. Cuando se utiliza una recta de regresión para la calibración, es adecuado utilizar sy/x, desviación detección, 124 estándar estimada, en lugar de sB en la estimación del límite de quedando la ecuación: LD = 3s y / x b ENSAYOS EN CÉLULAS Se prepara un cultivo de células endoteliales en PBS durante una hora. Por otro lado, se prepara una disolución de sonda 10-5 M en acetonitrilo y otra disolución de Hg(II) de concentración 10-5 M en PBS. A partir de estas disoluciones se preparan las muestras para realizar estos ensayos a la dilución indicada en cada caso. EXCÍMEROS Para estudiar la posible existencia de excímeros (dímeros excitados) se realiza un estudio de fluorescencia del producto variando su concentración. Comparando los espectros normalizados a diferentes concentraciones se puede observar si varía la forma de los mismos. INFLUENCIA DEL DISOLVENTE: En la cubeta de fluorescencia se introduce una cantidad de disolvente próxima a 2 ml. A este disolvente se añade una punta de espátula del compuesto objeto de estudio, obteniendo la disolución adecuada para el estudio de las propiedades ópticas. 124 J. N. Miller and J. C. Miller, Estadística y Químiometria para la Química Analítica Prentice Hall, Madrid, 2002. 3. Síntesis de derivados de triazinilglicina. Sensores 3.6. CONCLUSIONES - Se ha conseguido sintetizar nuevos derivados de glicinotriazina usando radiación microondas como fuente de energía. Además, se ha logrado completar las reacciones en tiempos cortos, con un método de purificación sencillo. La suma de todo ello hace que el método sintético diseñado sea medioambientalmente benigno. - Los derivados de N-triazinilglicina sintetizados no presentan un rendimiento cuántico de fluorescencia suficiente para su aplicación en dispositivos optoelectrónicos, sin embargo, se ha encontrado que interaccionan excelentemente con cationes metálicos. - Se ha estudiado el proceso de rotación restringida del enlace amino-triazina de la sal sódica del derivado de pirazol 13a. Se ha encontrado una temperatura de coalescencia de las señales de NH de 71 ºC. Con ello, se ha calculado la energía libre (∆G‡) de activación del proceso de rotación de 71,66 KJ mol-1. Este resultado viene a completar los estudios que previamente realizados por nuestro grupo para derivados sustituidos de 2,4-diaminotriazinas y ureido-1,3,5-triazinas. Los equilibrios de protonación de estas triazinilglicinas son complejos, por ello los espectros de RMN presentan diferentes desplazamientos químicos dependiendo del pH. Los estudios realizados al derivado de pirazol 13a, indican que debe poseer una estructura zwitteriónica por protonación del nitrógeno piridínico del anillo de triazina. Esta estructura justificaría el desapantallamiento de los NH, así como la dificultad para detectar todos los carbonos del anillo de triazina en medio ácido, que sí se observan en la sal sódica de este compuesto. - La elucidación de la estructura de los derivados de pirazol 13a y piperidina 13d por difracción de Rayos-X han confirmado la protonación del nitrógeno del anillo de 1,3,5-triazina. La estructura del derivado de pirazol en estado sólido muestra dos enlaces de hidrógeno intramoleculares entre los grupos NH y los anillos pirazólicos. El compuesto 195 196 3.6. Conclusiones se encuentra formando dímeros a través de enlaces de hidrógeno en los que están implicados un cloruro y un ácido carboxílico. Por otra parte, se observa que el grupo carboxilo se encuentra fuera del plano y está uniendo mediante interacciones débiles los dímeros, dando lugar a cadenas a lo largo del eje b. La unión de las cadenas, se ve reforzada por una interacción π−π, face-to-face, entre los anillos aromáticos unidos a la triazina. Es interesante destacar que el derivado de piperidina se encuentra formando dímeros a través de enlaces de hidrógeno entre los N y O de la glicina. Por otra parte, se observan interacciones π−π entre los anillos de triazina, con una geometría “offset-faceto-face”. - El derivado de naftilo 13g interacciona selectivamente con Hg(II), un catión muy dañino para el medio ambiente y de elevada toxicidad, con un límite de detección a nivel nanomolar, por debajo del nivel marcado por la EPA para contaminación en aguas. El método de Job nos revela que la interacción entre el ión y la sonda guarda una relación de 1 a 23 lo que podría indicar la formación de estructuras supramoleculares con huecos adecuados para albergar al ión Hg2+. El compuesto presenta una CCA de 1’9 x10-5 M y un radio hidrodinámico para la población mayoritaria de 217 nm, según las medidas de DLS en acetonitrilo. Por tanto, el derivado 13g es un excelente quimiosensor selectivo y altamente sensible para el catión Hg2+. Por otro lado, la N-triazinilglicina con sustituyente fenilo 13b interacciona selectivamente con Zn(II). El método de Job nos ha permitido establecer una estequiometria 1:1 para la interacción entre la sonda y el Zn2+. Los resultados obtenidos mediante espectroscopia de IR y RMN nos permiten proponer la coordinación del Zn(II) con los átomos de nitrógeno del anillo de triazina y con los oxígenos del grupo carboxilo, hipótesis que encajaría con el entorno tetraédrico habitual del ion Zn2+. En resumen, hemos sintetizado dos derivados de triazinilglicina que actúan como quimiosensores selectivos para los iones Hg(II) y Zn(II), respectivamente. Bibliografía 199 4. BIBLIOGRAFÍA Capítulo 1: 1. R. Breslau, Chemistry Today and Tomorrow. Awareness Chemical Society, Washington, 1997. 2. J. Clark, D. Macquarrie, Handbook of Green Chemistry & Technology, Blackwell Science, 2008. 3. World Commision of Environment and Development. Our Common Future. Oxford University Press. Oxford. 1987. 4. EPA (Environmental Protection Agency). 5. ACS (American Chemical Society). 6. P. T. Anastas, J. C. Warner. Green Chemistry. Theory and Practice. Oxford University Press. 1998. 7. Microwaves in Organic Synthesis, ed. A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012. 8. a) D. Stuerga, Microwaves in Organic Synthesis, Ed.: A. Loupy, A. de la Hoz, 3rd Edition, Wiley-VCH, Weinheim, 2012. b) M. D. P. Mingos MicrowaveAsssisted Organic Synthesis, Eds.: P. Lidström, J. P. Tierney, Blackwell, Oxford, 2005. 9. a) A. de la Hoz, A. Díaz-Ortiz, A. Moreno, Curr. Org. Chem., 2004, 8, 903. b) C. O. Kappe, Angew. Chem. Int. Ed., 2004, 43, 6250. c) M. Nüchter, B. Ondrushka, W. Bonrath, A. Gum, Green Chem., 2004, 6, 128. d) D. Bogdal, A. Loupy, Org. Proc. Res. & Dev., 2008, 12, 710. e) C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127. f) V. Polshettiwar, R. Varma, Chem. Soc. Rev., 2008, 37, 1546. g) C. O. Kappe, A. Stadler, D. Dallinger, Microwaves in Organic and Medicinal Chemistry, 2nd Ed. Wiley-VCH, Weinheim, 2012. 10. PD (Penetration Depth). 11. N. Elander, J.R. Jones, S. Y. Lu and S. Stone-Elander, Chem. Soc. Rev, 2000, 29, 239. 12. H. E. Blackwell, Org. Biomol. Chem., 2003, 1, 1251. 13. N. F. Kaiser, U. Bremberg, M. Larhed, C. Moberg and A. Hallberg. Angew. Chem. Int. Ed, 2000, 39, 3595. 14. D. R. Baghurst, D. M. P. Mingos, J. Chem Soc., Chem. Commun., 1992, 674. 200 Bibliografía 15. a) R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge, J. Rousell, Tetrahedron Lett., 1986, 27, 279. b) R. J. Giguere, T. L. Bray, S. M. Duncan, G. Majetich, Tetrahedron Lett., 1986, 27, 4945. 16. a) A. de la Hoz, A. Díaz-Ortiz, A. Moreno, Chem. Soc. Rev., 2005, 34, 164. b) A. de la Hoz, A. Díaz-Ortiz, A. Moreno, J. Microwave Power Electromagn. Energy, 2007, 41, 44. c) B. Pchelka, A. Loupy, A. Petit, Tetrahedron, 2006, 62, 10968. 17. M. A. Herrero, J. M. Kremsner, C. O. Kappe, J. Org. Chem., 2008, 73, 36. 18. D. Obermayer, B. Gutmann, C. O. Kappe, Angew. Chem. Int. Ed., 2009, 48, 44, 8321. 19. R. Banerjee, D. R. Brown, E. Weerapana, Synlett, 2013, 24, 1599-1605. 20. S. Kumar, H. R. Bhat, M. K. Kumawat, U. P. Singh, New J. Chem., 2013, 37, 581-584 y referencias allí citadas. 21. E. A. Peterson, P. S. Andrews, X. Be, A. A. Boezio, T. L. Bush, A. C. Cheng, J. R. Coats, A. E. Colletti, K. W. Copeland, M. DuPont, R. Graceffa, B. Grubinska, J.-C. Harmange, J. L. Kim, E. L. Mullady, P. Olivieri, L. B. Schenkel, M. K. Stanton, Y. Teffera, D. A. Whittington, T. Cai, D. S. La, Bioorg. Med. Chem. Lett., 2011, 21, 2064-2070. 22. a) S. Manohar, S. I. Khan, D. S. Rawat, Bioorg. Med. Chem. Lett., 2010, 322. b) H. R. Bhat, U. P. Singh, P. Gahtori, S. K. Ghosh, K. Gogoi, A. Prakash, R. K. Singhm, New J. Chem., 2013, 37, 2654-2662. 23. S. Nikolić, C. M. Keck, C. Anselmi, R. H. Müller, Int. J. Pharm., 2011, 414, 276-284. 24. http://www.espatentes.com/pdf/2188883_t3.pdf 25. M. J. Higuera Camacho, Tesis Doctoral, 2003, pp 7-19. Universidad de Córdoba. 26. A. I. Cañero, L. Cox, S. Redondo-Gómez, E. Mateos-Naranjo, M. C. Hermosín, J. Cornejo, J. Agric. Food Chem., 2011, 59, 5528-5534. 27. L. J. Krutz, D. L. Shaner, M. A. Weaver, R. M. T. Webb, R. M. Zablotowicz, K. N. Reddy, Y. Huang, S. J. Thomson, Pest. Manag. Sci., 2010, 66, 461-481. 28. T. J. Mooibroek, P. Gamez, Inorg. Chim. Acta, 360, 2007, 381. 29. B. Therrien, J. Organomet. Chem., 2011, 696, 637-651. Bibliografía 201 30. F. Gärtner, D. Cozzula, S. Losse, A. Boddien, G. Anilkumar, H. Junge, T. Schulz, N. Marquet, A. Spannenberg, S: Gladiali, M. Beller, Chem. Eur. J., 2011, 17, 6998-7006. 31. a) P. Gamez, J. Reedijk, Eur. J. Inorg. Chem., 2006, 29; b) R. Wang, C. Pellerin, O. Lebel, J. Mater. Chem., 2009, 19, 2747; c) F. Vera, J. Barberá, P. Romero, J. L. Serrano, M. B. Ros, T. Sierra, Angew. Chem. Int. Ed., 2010, 49, 4910; d) J. Barber, L. Puig, P. Romero, J. L. Serrano, T. Sierra, J. Am. Chem. Soc., 2005, 127 (1), 458; e) A. Delori, E. Suresh, V. R. Pedireddi, Chem. Eur. J., 2008, 14, 6967; f) T. Seki, S. Yagai, T. Karatsu, A. Kitamura, J. Org. Chem., 2008, 73 (9), 3328; g) S. Yagai, S. Kubota, K. Unoike, T. Karatsu, A. Kitamura, Chem. Commun., 2008, 4466. 32. G. M. Whitesides, J. P. Mathias, C. T. Seto, Science, 1991, 254, 1312. 33. W. J. Qi, D. Wu, J. Ling, C. Z. Huang, Chem. Commun., 2010, 46, 4893. 34. J. D. Wuest, A. Rochefort, Chem. Commun., 2010, 46, 2923. 35. B. K. Mishra, J. S. Arey, N. Sathyamurthy, J. Phys. Chem. A, 2010, 114, 96069616. 36. Y. Hisamatsu, H. Aihara, Chem. Commun. 2010, 46, 4902. 37. T. Kawamichi, T. Haneda, M. Kawano, M. Fujita, Nature, 2009, 461, 633. 38. S. Ren, D. Zeng, H. Zhong, Y. Wang, S. Qian, Q. Fang, J. Phys. Chem. B, 2010, 114, 10374. 39. A. Richard, H. A. Klenklera, A. Tranc, D. Z. Popovic, G. Xu, Org. Elect. 2008, 9, 285. 40. M. M. Rothmann, S. Haneder, E. Da Como, C. Lennartz, C. Schildknetch, P. Strohriegl, Chem. Mater., 2010, 22, 2403. 41. J. Pang, Y. Tao, S. Frieberg, X-P. Yang, M. D’iorio, S. Wang. J. Mater. Chem. 2002, 12, 206. 42. Y. Jiang, Y. Wang, B. Wang, J. Yang, N. He, S. Qian, J. Hua, Chem. Asian J., 2011, 6, 157-165 y referencias citadas en la nota 1 de este artículo. 43. J. Liu, K. Wang, X. Zhang, C. Li, X. You, Tetrahedron, 2013, 69, 190-200. 44. M. A. Özdağ, T. Ceyhan, H. G. Yaglioglu, A. Elmali, Ö. Bekaroğlu, Opt. & Laser Tech., 2011, 992-995. 45. DSSCs (Dye Sensitized Solar Cells). 46. J. Liu, K. Wang, F. Xu, Z. Tang, W. Zheng, J. Zhang, C. Li, T. Yu, X. You, Tetrahedron Letters, 2011, 52, 6492. y referencias citadas. 202 Bibliografía 47. a) X. Cheng, J. Jin, Q. Li, X. Dong, Chin. J. Chem., 2010, 28, 1957-1962. b) H. K. Dambal, C. V. Yelamaggad, Tetrahedron Letters, 2012, 53, 186-190. 48. M. Tatina, S. K. Yousuf, D. Mukherjee, Org. Biomol. Chem., 2012, 10, 53575360. 49. E. A. Prasetyanto, M. B. Ansari, B.-H. Min, S.-E. Park, Catalysis today, 2010, 252-257. 50. D. Usachov, O. Vilkov, A. Grüneis, D. Haberer, A. Fedorov, V. K. Adamchuk, A. B. Preobrajenski, P. Dudin, A. Barinov, M. Oehzelt, C. Laubschat, D. V. Vyalikh, Nano Lett., 2011, 11, 5401-5407. 51. V. León, M. Quintana, M. A. Herrero, J. L. G. Fierro, A. de la Hoz, M. Prato, E. Vázquez, Chem. Commun., 2011, 47, 10936-10938. 52. L. Xiong, Z. He, H. Xu, J. Lu, T. Ren, X. Fu, Lubr. Sci., 2011, 23, 33-40. 53. K. Yang, Y. H. Park, S. G. Cho, H. W. Lee, C. K. Kim, H.-J. Koo, J. Comput. Chem., 2010, 31, 2483-2492. 54. H. Lim, M. C. Cha, J. Y. Chang, Macromol. Chem. Phys., 2012, 213, 13851390. Capítulo 2: 55. a) L. D. S. Yadav, R. Kapoor, Tetrahedron, 2003, 44, 8951 b)L. D. S. Yadav, S. Yadav, V. K. Rai, Green Chem., 2006, 8, 455. c) L. D. S. Yadav, V. K. Rai, S. Yadav, Lett. Org. Chem., 2007, 4, 47. 56. A. Dandia, K. Arya, M. Sati, P. Sarawgi, J. Fluor. Chem., 2004, 125, 1273. 57. A. Diaz-Ortíz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, M. Mateo, A. Sánchez-Migallón, G. Valiente, New. J. Chem., 2004, 28, 952. 58. B. R. Manzano, F. A. Jalón, M. L. Soriano, M. C. Carrión, M. P. Carranza, K. Mereiter, A. M. Rodríguez, A. de la Hoz, A. Sánchez-Migallón, Inorg. Chem. 2008, 47, 8957. 59. B. R. Manzano, F. A. Jalón, M. L. Soriano, A. M. Rodríguez, A. de la Hoz, A. Sánchez-Migallón, Cryst. Growth Des., 2008, 8, 5, 1585. 60. J-J. Shie, J-M. Fang, J. Org. Chem. 2007, 72, 3141. 61. H. Chen, P. Dao, A. Laporte, C. Garbay, Tetrahedron, 2010, 51, 3174. 62. A. Díaz-Ortiz, A. de la Hoz, A. Moreno, A. Sánchez-Migallón, G. Valiente, Green Chem., 2002, 4, 339. 63. K. Arya, A. Dandia, Bioorg. Med. Chem. Let. 2007, 17, 3298. Bibliografía 203 64. A. Diaz-Ortíz, J. Elguero, A. de la Hoz, A. Jiménez, A. Moreno, S. Moreno, A. Sánchez-Migallón, QSAR Comb. Sci., 2005, 24, 649. 65. K. Doktorov, V. B. Kurteva, D. Ivanova, I. Timtcheva, ARKIVOC, 2007, XV, 232. 66. A. Diaz-Ortíz, J. Elguero, C. Foces-Foces, A. de la Hoz, A. Moreno, S. Moreno, A. Sánchez-Migallón, G. Valiente, Org. Biomol. Chem., 2003, 1, 4451. 67. M. Moral, A. Ruiz Carretero, M. I. López Solera, A. Sánchez-Migallón, A. de la Hoz. Tetrahedron, 2010, 66, 121-127. 68. A. Ruiz Carretero, J. R. Ramírez, A. Sánchez-Migallón, A. de la Hoz, Eur. J. Org. Chem, submitted manuscript. 69. E. Beltrán, J. L. Serrano, T. Sierra, R. Giménez, Org. Lett. 2010, 12, 1404. 70. El desplazamiento de Stokes se define como la diferencia de energía entre la longitud de onda más intensa en el espectro de absorción y la longitud de onda más intensa en el espectro de emisión. 71. Se considera que valores de Stokes por debajo de 5000 cm-1 corresponden a una pérdida de energía típica de relajaciones rotacionales y vibracionales. 72. B. Valeur, “Molecular fluorescence. Principles and Applications”, Wiley-VCH, Weinheim (Alemania), 2002. 73. A. García, B. Insuasty, M. A. Herranz, R. Martínez-Álvarez, N. Martín, Org. Lett. 2009, 11, 5398. 74. En física teórica, “cut-off” es el valor máximo o mínimo de energía, momento o longitud, que nos indica que los objetos cuyas propiedades físicas queden fuera de esos límites serán ignorados. El “cut-off” en ultravioleta es la máxima energía permitida o la longitud de onda más pequeña permitida. 75. A. P. H. J. Schenning, P. Jonkheijm, E. Peeters, E. W. Meijer, J. Am. Chem. Soc. 2001, 123, 409. 76. (a)Y. B. Lim, E. Lee, M. Lee, Angew. Chem. Int. Ed., 2007, 46, 9011-9014. (b)M. C. A. Stuart, J. C. van de Pas, J. Engberts, J. Phys. Org. Chem. 2005, 18, 929-934. 77. http://en.wikipedia.org/wiki/Nile_red 78. DOSY (Diffusion Ordered Spectroscopy). 79. M. El-Sedik, N. Almonasy, M. Nepraš, S. Bureš, M. Dvořák, M. Michl, J. Čermak, R. Hrdina, Dyes Pigments, 2012, 92, 1126-1131. 204 Bibliografía 80. J. Rodriguez, J. Gonzalo, J. L. Tejedor, J. Org. Chem., 2002 , 67, 22, 76317640. 81. W. Wei, H-J. Wang, C-Q. Jiang, Spectrochimica Acta Part A, 2008, 70, 362366. 82. R. M. Desai, D. K. Dodiya, A. R. Trivedi, V. H. Shah, Med. Chem. Res., 2008, 17, 495-506. 83. CAC (Concentración de Agregación Crítica). Capítulo 3: 84. C. Rubio, D. González Weller, R. E. Martín-Izquierdo, C. Revert, I. Rodríguez y A. Hardison, Nutr. Hosp., 2007, 22, 1, 101-107 y referencias allí citadas. 85. Real Decreto 1138/90, de 14 de septiembre, por el que se aprueba la Reglamentación Técnico-Sanitaria para el abastecimiento y control de las aguas potables de consumo público. BOE (226):27488-97. 86. M. B. Salzman, E. M. Smith, C. Koo. Excessive oral zinc supplementation. J. Pediatr. Hematol. Oncol., 2002; 24, 7, 582-584. 87. X. Gaona Martínez, Tesis Doctoral. 2004. pp 9-49. Universidad Autónoma de Barcelona. 88. R. P. Mason, W. F. Fitzgerald, F. M. M. Morel, Geochim. Cosmochim. Acta, 1994, 58, 3191-3198. 89. M. Santra, B. Roy, K. H. Ahn, Org. Lett., 2011, 13, 13, 3422-3425. 90. J. Du, M. Hu, J. Fan, X. Peng., Chem. Soc. Rev., 2012, 41, 4511-4535. 91. a) B. T. Nguyen, E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118-3127. b) L. A. Cabell, M. D. Best, J. J. Lavigne, S. E. Schneider, D. M. Perreault, M.-K. Monahan, E. V. Anslyn, J. Chem. Soc.,Perkin Trans. 2, 2001, 315-323. c) A. R. Ray, Trends Neurosci., 2006, 29, 200-206. 92. K. Kaur, R. Saini, A. Kumar, V. Luxami, N. Kaur, P. Singh, S. Kumar, Coord. Chem. Rev., 2012, 256, 1992-2028. 93. M.Y. Chae, A.W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704-9705. 94. M.-H. Yang, P. Thirupathi, K.-H. Lee, Org. Lett., 2011, 13, 19, 5028-5031. 95. J. Du, S. Yin, L. Jiang, B. Ma, X. Chen, Chem. Commun., 2013, 49, 4196-4198. Bibliografía 205 96. P. Srivastava, R. Ali, S. S. Razi, M. Shahid, S. Patnaik, A. Misra, Tetrahedron Letters, 2013, 54, 3688-3693. 97. K. Ghosh, T. Sarkar, A. Samadder, A. R. Khuda-Bukhsh, New. J. Chem, 2012, 36, 2121-2127. 98. M. Sellaiah, Y. C. Rajan, H.-C. Lin, J. Mater. Chem, 2012, 22, 8976-8987. 99. M. Kumar, N. Kumar, V. Bhalla, Chem. Commun., 2013, 49, 877-879. 100. K. Baek, M. S. Eom, S. Kim, M. S. Han, Tetrahedron Letters, 2013, 54, 16541657. 101. Y. Cai, X. Meng, S. Wang, M. Zhu, Z. Pan, Q. Guo, Tetrahedron Letters, 2013, 54, 1125-1128. 102. a) G. Blotny, Tetrahedron, 2006, 62, 9507. b) J. A. Zerkowski, J. P. Mathias, G. M. Whitesides, J. Am. Chem. Soc. 1994, 116, 4305. 103. P. Kociensky, Protecting Groups, 3rd Ed.; Thieme Verlag: Stuttgart, 2006. 104. E. Hollink, E. E. Simanek, D. E. Bergbreiter, Tetrahedron Letters, 2005, 46, 2005-2008. 105. A. de la Hoz, A. Sánchez-Migallón, B. T. Pelado, J. R. Ramírez, resultados sin publicar. 106. J. Sandström, Dynamic NMR Spectroscopy. Academic Press: New York, 1982, 96. 107. S. M. S. Chauhan, N. G. Giri, Supramolecular Chemistry, 20, 8, 2008, 743-752. 108. V. Kampyli, D. A. S. Phillips, A. H. M. Renfrew, Dyes and Pigments, 2004, 165-175. 109. δ NH2 melamina = 6’2 ppm.. 110. B. Wang and E. J. Anslyn, Chemosensors: Principles, Strategies, and Applications, John Wiley and Sons, New York, 2011. 111. Aclaración. 112. a) IUPAC. Compendium of Chemical Terminology. 2nd Ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications. Oxford. 1997. b) International Union of pure and Applied Chemistry. Nomenclature, Symbols, Units and Their Usage in Spectrochemical Analysis. 2. Data Interpretation. Spectrochimica Acta. 33, 242 (1978). c) J.N Miller and J. C. Miller, Estadística y Químiometria para la Química Analítica Prentice Hall, Madrid, 2002. 206 Bibliografía 113. Mercury Update: Impact of Fish Advisories. EPA Fact Sheet EPA-823-F-01011; EPA, Office of Water: Washington, DC, 2001. 114. Algunas revisiones recientes: a) D. T. Quang, J. S. Kim, Chem. Rev., 2010, 110, 6280; b) X. Chen, X. Tian, I. Shin, J. Yoon, Chem. Soc. Rev., 2011, 40, 4783; c) A. Razgulin, N. Ma, J. Rao, Chem. Soc. Rev., 2011, 40, 4186; d) Z. Liu, W. He, Z. Guo, Chem. Soc. Rev., 2013, 42, 1568-1600. 115. P. Job, Ann. Chim.-Rome Appl., 1928, 9, 113-203. 116. Ensayos realizados con la colaboración del Dr. Mario Durán, profesor de la Facultad de Medicina. 117. K. A. Kolmakov, J. Heterocyclic Chem., 2008, 45, 533-539. 118. W. Karuehanon, W. Fanfuenha, A. Rujiwatra, M. Pattarawarapan, Tetrahedron Letters, 2012, 53, 3486-3489. 119. R. F. Chen, Arch. Biochem. Biophys., 1971, 142, 2, 552–564. 120. S. E. Bryan, A. L. Guy, K. J. Hardy, Biochemistry-US, 1974, 13, 2, 313–319. 121. a) Z.-K. Song, B. Dong, G.-J. Lei, M.-J. Peng, Y. Guo, Tetrahedron Lett., 2013, 54, 4045-4949 b) K. Tsukamoto, S. Iwasaki, M. Isaji, H. Maeda, Tetrahedron Lett., 54, 5971-5973. 122. J. A. Zerkowski, J. C. MacDonald, C. T. Seto, D. A. Wierda, G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 2382. 123. F. H. S. Curd, J. K. Landquist, F. L. Rose, J. Chem. Soc., 1947, 154. 124. J. N. Miller and J. C. Miller, Estadística y Químiometria para la Química Analítica. Prentice Hall, Madrid, 2002. Anexo 1. 12 principios de la química sostenible. 209 ANEXO 1. 12 PRINCIPIOS DE LA QUÍMICA SOSTENIBLE: 6 1. Prevención: Es preferible evitar la producción de un residuo que tratar de limpiarlo una vez que se haya formado. 2. Economía atómica: Los métodos de síntesis deberán diseñarse de manera que incorporen al máximo, en el producto final, todos los materiales usados durante el proceso, minimizando la formación de subproductos. 3. Uso de metodologías que generen productos con toxicidad reducida: Siempre que sea posible, los métodos de síntesis deberán diseñarse para utilizar y generar sustancias que tengan poca o ninguna toxicidad, tanto para el hombre como para el medio ambiente. 4. Generar productos eficaces pero no tóxicos: Los productos químicos deberán ser diseñados de manera que mantengan la eficacia a la vez que reduzcan su toxicidad. 5. Reducir el uso de sustancias auxiliares: Se evitará, en lo posible, el uso de sustancias que no sean imprescindibles (disolventes, reactivos para llevar a cabo separaciones, etc.) y en el caso de que se utilicen que sean lo más inocuos posible. 6. Disminuir el consumo energético: Los requerimientos energéticos serán catalogados por su impacto medioambiental y económico, reduciéndose todo lo posible. Se intentará llevar a cabo los métodos de síntesis a temperatura y presión ambientes. 7. Utilización de materias primas renovables: La materia prima ha de ser preferiblemente renovable en vez de agotable, siempre que sea técnica y económicamente viable. 6 P. T. Anastas, J. C. Warner. Green Chemistry. Theory and Practice. Oxford University Press. 1998 210 8. Evitar la derivatización innecesaria: Se evitará en lo posible la formación de derivados (grupos de bloqueo, de protección y desprotección, modificación temporal de procesos físicos y químicos). 9. Potenciación de la catálisis: Se emplearán catalizadores (lo más selectivos posible), reutilizables en lo posible, en lugar de reactivos estequiométricos. 10. Generar productos biodegradables: Los productos químicos se diseñarán de tal manera que al finalizar su función no persistan en el medio ambiente sino que se transformen en productos de degradación inocuos. 11. Desarrollar metodologías analíticas para la monitorización en tiempo real: Las metodologías analíticas serán desarrolladas posteriormente para permitir una monitorización y control en tiempo real del proceso, previo a la formación de sustancias peligrosas. 12. Minimizar el potencial de accidentes químicos: Se elegirán las sustancias empleadas en los procesos químicos de forma que se minimice el riesgo de accidentes químicos, incluidas las emanaciones, explosiones e incendios. Anexo 2. Espectros. 211 ANEXO 2: ESPECTROS. En este anexo se han incluido los espectros más representativos de los productos sintetizados. 1) 2,5-DIMETOXIFENILAMINOTRIAZINAS: N-(2,5-Dimetoxifenil)-N’,N’’-bis-(2-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (3a). Espectro de 1H-RMN de 3a (DMSO, 25 ºC). N N O H N N N O NH N HN N N 3a 212 Espectro de 1H-RMN de 3a (DMSO, 80 ºC): N N O H N N N 13 N HN O Espectro de NH N N 3a C-RMN de 3a (DMSO, 80 ºC): N N O H N N N O NH N HN N N 3a Anexo 2. Espectros. 213 Espectro de IR de 3a (neto): 100 %T 3398,57 98 1049,28 N N 96 O H N 1217,08 NH N 1575,84 N 94 N 1556,55 O HN N N 92 1506,41 3a 1417,68 90 88 3500 JR3a 3000 2500 2000 1750 1500 1250 1000 750 1/cm Espectro de UV, fluorescencia y excitación de 3a (CH2Cl2, 10-5 M): 3,0 UV Fluorescencia Excitación 340 nm Excitación 398 nm 0,8 2,0 N N O H N N N 0,4 O NH N 1,5 HN N N 1,0 3a 0,2 0,5 0,0 0,0 250 300 350 400 450 Longitud de onda (nm) 500 550 600 Intensidad (a. u.) Absorbancia 0,6 2,5 214 Distribución de diámetros hidrodinámicos en DLS de una disolución de 3a en THF a una concentración de 10-3M: Anexo 2. Espectros. 215 N-(2,5-Dimetoxifenil)-N’,N’’-difenil-1,3,5-triazina-2,4,6-triamina (3b). Espectro de 1H-RMN de 3b (DMSO, 80 ºC): O H N NH N N O N HN 3b Espectro de 13C-RMN de 3b (DMSO, 80 ºC): O H N N N O HN 3b NH N 216 Espectro de IR de 3b (neto): 100 %T 92,5 1230,58 1579,70 1556,55 95 1049,28 1024,20 1633,71 3392,79 97,5 754,17 1417,68 1398,39 87,5 688,59 1514,12 90 85 82,5 3500 JR4a 3000 2500 2000 1750 1500 1250 1000 750 1/cm Espectro de UV, fluorescencia y excitación de 3b (CH2Cl2, 10-5 M): 0,8 UV Fluorescencia Excitación 1,8 1,6 0,6 1,4 0,5 1,2 1,0 0,4 0,8 0,3 0,6 0,2 0,4 0,1 0,0 250 0,2 300 350 400 450 Longitud de onda (nm) 500 550 0,0 600 Intensidad (a. u.) Absorbancia 0,7 Anexo 2. Espectros. 217 Distribución de diámetros hidrodinámicos en DLS de una disolución 10-2 M de 3b en CH2Cl2: O H N N N O HN 3b NH N 218 N-(2,5-Dimetoxifenil)-N’,N’’-bis-(p-metoxifenil)-1,3,5-triazina-2,4,6-triamina (3c). Espectro de 1H-RMN de 3c (DMSO, 80 ºC): O H N NH N N OCH3 N HN O 3c OCH3 Espectro de 13C-RMN de 3c (DMSO, 80 ºC): O H N N N NH OCH3 N HN O 3c OCH3 Anexo 2. Espectros. 219 Espectro de IR de 3c (neto): 100 %T 3419,79 3394,72 95 90 796,60 823,60 1246,02 1213,23 1479,40 1409,96 80 1026,13 85 1512,19 75 70 3500 JR5a 3000 2500 2000 1750 1500 1250 1000 750 1/cm Espectro de UV, fluorescencia y excitación de 3c (CH2Cl2, 10-5 M): 0,30 5 UV Fluorescencia Excitación 0,25 4 3 0,15 2 0,10 1 0,05 0,00 250 300 350 400 450 Longitud de onda (nm) 500 550 0 600 Intensidad (a. u.) Absorbancia 0,20 220 N-(2,5-Dimetoxifenil)-N’,N’’-bispiperidino-1,3,5-triazina-2,4,6-triamina (3d). Espectro de 1H-RMN de 3d (DMSO, 80 ºC): O H N N N N N N O 3d Espectro de 13C-RMN de 3d (DMSO, 80 ºC): O H N N N N N O 3d N Anexo 2. Espectros. 221 Espectro de IR de 3d (neto): 100 %T 2933,73 2848,86 95 90 75 1504,48 70 773,46 1589,34 1489,05 1456,26 1435,04 1537,27 80 1284,59 1251,80 85 65 3500 JR6a 3000 2500 2000 1750 1500 1250 1000 750 1/cm Espectro de UV, fluorescencia y excitación de 3d (CH2Cl2, 10-5 M): 2,5 UV Fluorescencia 259 nm Fluorescencia 305 nm Excitación 340 nm Excitación 405 nm 0,25 2,0 1,5 0,15 1,0 0,10 0,5 0,05 0,00 300 400 Longitud de onda (nm) 500 0,0 600 Intensidad (a. u.) Absorbancia 0,20 222 Distribución de diámetros hidrodinámicos en DLS de una disolución de 3d en THF de concentración 10-3 M. Anexo 2. Espectros. 223 N-(2,5-Dimetoxifenil)-N’,N’’-bismorfolino-1,3,5-triazina-2,4,6-triamina (3e). Espectro de 1H-RMN de 3e (DMSO, 25ºC): O O H N N N N N N O 3e O Espectro de 13C-RMN de 3e (DMSO, 25ºC): O O H N N N N O N N 3e O 224 Espectro de IR de 3e (neto): 100 95 3414,00 %T 800,46 1114,86 1506,41 75 1255,66 80 1471,69 1444,68 1427,32 1531,48 85 1215,15 1359,82 1575,84 90 70 3500 JR7a 3000 2500 2000 1750 1500 1250 1000 750 1/cm Espectro de UV, fluorescencia y excitación de 3e (CH2Cl2, 10 -5 M): 3,0 UV Fluorescencia 259 nm Fluorescencia 305 nm Excitacion 340 nm 0,7 0,6 2,5 2,0 0,4 1,5 0,3 1,0 0,2 0,5 0,1 0,0 0,0 300 400 Longitud de onda (nm) 500 600 Intensidad (a. u.) Absorbancia 0,5 Anexo 2. Espectros. 225 Distribución de diámetros hidrodinámicos de las especies en una disolución de 3e en CH2Cl2 de concentración 10-2M. 226 2) MONOTRIAZINAS CON NAFTALENO: N-4,6-(Di(piperidin-1-il)-1,3,5-triazin-2-il)naftaleno-1,5-diamina (5d): Espectro de 1H-RMN de 5d (DMSO, 25ºC): H N H 2N N N N 5d Espectro de 13C-RMN de 5d (DMSO, 25ºC): H N H 2N N N N N 5d N N N Anexo 2. Espectros. 227 Espectro de IR de 5d (neto): Espectro de UV, fluorescencia y excitación de 5d (CH2Cl2, 10 -5 M): 4 0,6 UV Fluorescencia 234 nm Fluorescencia 335 nm Excitación 400 nm Excitación 428 nm 0,5 3 Absorbancia 0,3 2 0,2 1 0,1 0,0 0 300 400 Longitud de onda (nm) 500 600 Intensidad (a. u.) 0,4 228 3) NAFTALENO BISTRIAZINAS: N2,N2'-(Naftaleno-1,5-diil)bis(N4,N6-difenil-1,3,5-triazina-2,4,6-triamina) (6b). Espectro de 1H-RMN de 6b (DMSO, 80 ºC): NH N N N N H NH HN 6b H N N N N HN Espectro de 13C-RMN de 6b (DMSO, 80 ºC): NH N N H N NH N HN 6b H N N N HN N Anexo 2. Espectros. 229 Espectro de IR de 6b (neto): %T 3439,08 99 1479,40 806,25 688,59 1494,83 1514,12 93 1352,10 1573,91 94,5 750,31 1556,55 96 1228,66 1625,99 775,38 97,5 91,5 1440,83 90 1392,61 88,5 87 3600 JR-29 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectro de UV, fluorescencia y excitación de 6b (CH2Cl2, 10-6 M). UV Fluorescence 268 nm Fluorescence 323 nm Excitation 200 0,6 150 0,4 100 0,2 50 0,0 300 400 Wavelength (nm) 0 500 Intensity (a.u.) Absorbance 0,8 230 Espectros de UV normalizados en diferentes disolventes de 6b. UV Hexane UV DCM UV CH3CN 1,0 UV MeOH NH N Absorbance N H N N NH 6b H N N HN 0,5 N N HN 0,0 200 300 400 Wavelength (nm) Espectros de fluorescencia normalizados en diferentes disolventes de 6b. Fluorescence Hexane Fluorescence DCM Fluorescence CH3CN 1,0 Fluorescence MeOH Intensity (a.u.) 0,8 NH N 0,6 N H N NH N 0,4 6b 0,2 0,0 300 H N N HN N N HN 400 Wavelength (nm) 500 Anexo 2. Espectros. 231 Distribución de diámetros hidrodinámicos de partículas de 6b en una disolución de concentración 8x10-4 M en CH2Cl2. 120 Normalized Counts DiPh_800 uM_DCM 100 NH 80 N N H 60 40 NH N 20 H N N HN 6b 0 N N N HN 0 50 100 150 200 250 300 350 Hydrodynamic Diameter (nm) 232 N2,N2'-(Naftaleno-1,5-diil)bis(N4,N6-bis(4-metoxifenil)-1,3,5-triazina-2,4,6triamina) (6c). Espectro de 1H-RMN de 6c (DMSO, 25 ºC): O NH O N N H N N NH HN H N N N N O HN 6c O Espectro de 1H-RMN de 6c (DMSO, 80 ºC): O NH O N N H N N NH HN N 6c H N N N O HN O Anexo 2. Espectros. 233 Espectro de 13C-RMN de 6c (DMSO, 80 ºC): Espectro de IR de 6c (neto): 3342,64 99 779,24 1415,75 1556,55 94,5 825,53 1172,72 96 1026,13 1604,77 97,5 1296,16 3406,29 %T 1487,12 1392,61 91,5 1232,51 1384,89 93 90 3600 JR-24 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm 234 Espectro de UV, fluorescencia y excitación de 6c (CH2Cl2, 10-6 M). 50 UV Fluorescence Excitation 0,6 0,5 O N N H N N NH 0,3 HN H N N N 0,2 Intensity (a. u.) Absorbance NH O 0,4 N O HN 6c O 0,1 0,0 300 0 500 400 Wavelength (nm) Espectros de UV normalizados en diferentes disolventes de 6c. UV Hexane UV DCM UV CH3CN O UV MeOH NH O 1,0 N Absorbance N H N N NH HN N 0,5 6c H N N N O HN O 0,0 200 300 Wavelength (nm) 400 Anexo 2. Espectros. 235 Espectros de fluorescencia normalizados en diferentes disolventes de 6c. Fluorescence Hexane Fluorescence DCM Fluorescence CH3CN 1,0 Fluorescence MeOH O Intensity (a. u.) 0,8 NH O N N H 0,6 0,4 N N NH N 6c 0,2 H N N HN N O HN O 0,0 300 400 Wavelength (nm) 500 236 - N1,N5-Bis(4,6-di(piperidin-1-il)-1,3,5-triazin-2-il)naftaleno-1,5-diamina (6d). Espectro de 1H-RMN de 6d (CDCl3, 25ºC): N N N N N NH HN N N 6d N N N Espectro de 13C-RMN de 6d (CDCl3, 25ºC): N N N N N NH HN N N 6d N N N Anexo 2. Espectros. 237 Espectro de IR de 6d (neto): N N N N NH N HN N 6d N N N N Espectro de UV, fluorescencia y excitación de 6d (CH2Cl2, 10-6 M). UV Fluorescence 237 nm Fluorescence 336 nm Excitation 1,0 350 300 0,8 0,6 200 150 0,4 100 0,2 50 0,0 300 400 Wavelength (nm) 0 500 Intensity Absorbance 250 238 Espectros UV normalizados en diferentes disolventes de 6d. UV DCM UV CH3CN 1,0 UV MeOH UV Hexane Absorbance 0,8 N N 0,6 N N NH N 0,4 HN 0,2 0,0 200 N N 6d N N N 300 400 Wavelength (nm) Espectros de fluorescencia normalizados en diferentes disolventes de 6d. Fluorescence DCM Fluorescence CH3CN 1,0 Fluorescence MeOH Fluorescence Hexane Intensity (a.u.) 0,8 N N 0,6 N N N NH 0,4 HN N N 6d 0,2 0,0 300 400 Wavelength (nm) N N N 500 Anexo 2. Espectros. 239 Distribución de diámetros hidrodinámicos de 6d en una disolución de concentración 2 x 10-4 M en CH2Cl2. Normalized Counts _0.2 mM_DCM 40 N N 30 N N N NH 20 HN 10 0 N N 6d 0 20 40 60 N N N 80 Hydrodynamic Diameter (nm) 100 240 N1,N5-Bis(4,6-dimorfolino-1,3,5-triazin-2-il)naftaleno-1,5-diamina (6e). Espectro de 1H-RMN de 6e (CDCl3, 25ºC): O N N N N N NH O O HN 6e N N N N O Espectro de 13C-RMN de 6e (CDCl3, 25ºC): O N N N N N NH O O HN 6e N N N N N O N Anexo 2. Espectros. 241 Espectro de IR de 6e (neto): 100 %T 99,9 99,8 1010,70 800,46 858,32 99,7 1394,53 1539,20 2900,94 2980,02 99,6 1516,05 2362,80 99,4 1118,71 1597,06 3464,15 99,5 1253,73 99,3 99,2 3600 JR-25 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectro de UV, fluorescencia y excitación de 6e (CH2Cl2, 10-6 M): 700 1,0 UV Fluorescence 236 nm Fluorescence 336 nm Excitation 0,8 600 0,6 400 300 0,4 200 0,2 100 0,0 300 400 Wavelength (nm) 0 500 Intensity (a. u.) Absorbance 500 242 Espectros UV de 6e normalizados en diferentes disolventes. UV Hexane UV DCM UV CH3CN O N 1,0 N N Absorbance 0,8 UV MeOH N N NH O O HN 0,6 6e N N N N N 0,4 O 0,2 0,0 200 300 400 Wavelength (nm) Espectros de fluorescencia normalizados de 6e en diferentes disolventes. Fluorescence Hexane Fluorescence DCM Fluorescence CH3CN 1,0 Fluorescence MeOH O Intensity (a.u.) 0,8 N N N 0,6 N N NH O 0,4 O HN 6e 0,2 N N N N N O 0,0 400 500 Wavelength (nm) Anexo 2. Espectros. 243 N2,N2'-(Naftaleno-1,5-diil)bis(N4,N4,N6,N6-tetrafenil-1,3,5-triazina-2,4,6-triamina) (6f). Espectro de 1H-RMN de 6f (DMSO, 80 ºC): N N HN N N N NH 6f N N Espectro de 13C-RMN de 6f (DMSO, 80 ºC): N N HN N N N N N N NH N N N 6f N N 244 Espectro de IR de 6f (neto): 100 %T N N N NH 6f N 744,52 846,75 1537,27 80 1390,68 N 1504,48 N 796,60 779,24 N N HN 1168,86 1587,42 N 85 1328,95 1309,67 90 1028,06 3230,77 3095,75 95 1546,91 N 1236,37 75 70 3600 JR-27 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectro de UV, fluorescencia y excitación de 6f (CH2Cl2 , 10-6 M): 800 1,0 UV Fluorescence 244 nm Fluorescence 336 nm Excitation 0,8 700 500 0,6 400 0,4 300 200 0,2 100 0,0 200 300 400 Wavelength (nm) 0 500 Intensity (a.u.) Absorbance 600 Anexo 2. Espectros. 245 Espectros UV normalizados en diferentes disolventes de 6f. UV Hexane UV DCM UV CH3CN 1,0 N N Absorbance 0,8 HN 0,6 N N N UV MeOH N N N NH N 6f N 0,4 0,2 0,0 200 300 400 500 Wavelength (nm) Espectros de fluorescencia normalizados en diferentes disolventes de 6f. Fluorescence Hexane Fluorescence DCM Fluorescence CH3CN 1,0 Fluorescence MeOH Intensity (a.u.) 0,8 N N HN N N 0,6 N N N 0,4 NH N 6f N 0,2 0,0 300 400 Wavelength (nm) 500 N 246 N2,N2'-(Naftaleno-1,5-diil)bis(N4,N6-bis(4-nitrofenil)-1,3,5-triazina-2,4,6-triamina) (6h). Espectro de 1H-RMN de 6h (DMSO, 80 ºC): O 2N NH O2 N N N N H N NH HN 6h H N N N N NO2 HN NO2 Espectro de 13C-RMN de 6h (DMSO, 80 ºC): O 2N NH O2 N N N H N N NH HN 6h H N N N N NO2 HN NO2 Anexo 2. Espectros. 247 Espectro de IR de 6h (neto): 100 HN N N NO2 HN 90 1111,00 H N N 798,53 1230,58 NH 92 6h 1249,87 N N 1409,96 N H 1504,48 1485,19 N 94 750,31 NH O2N 1591,27 1562,34 1622,13 O2N 846,75 96 1182,36 3388,93 98 3327,21 %T 1301,95 NO2 1323,17 88 86 3600 JR-26 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectro de UV, fluorescencia y excitación de 6h (CH2Cl2, 10-5 M): UV Fluorescence 238 nm Fluorescence 355 nm Excitation 387 nm Excitation 436 nm 0,30 0,25 50 40 O2N O2N N N H 30 N N NH 0,15 HN 6h H N N N 20 N NO2 HN 0,10 NO2 10 0,05 0,00 200 300 400 500 Wavelength (nm) 600 0 700 Intensity (a.u.) Absorbance NH 0,20 248 Espectros de UV normalizados en distintos disolventes de 6h. UV Hexane UV DCM UV CH3CN O 2N 1,0 UV MeOH NH O2 N Absorbance 0,8 N N H N N NH HN 0,6 6h H N N N N NO2 HN 0,4 NO2 0,2 0,0 200 300 400 500 600 Wavelength (nm) Espectros de fluorescencia normalizados con distintos disolventes de 6h. Fluorescence Hexane Fluorescence DCM Fluorescence CH3CN Fluorescence MeOH 1,0 O 2N NH O2 N Intensity (a.u.) 0,8 N N H N N NH 0,6 HN 6h H N N N N NO2 HN 0,4 NO2 0,2 0,0 300 400 Wavelength (nm) 500 Anexo 2. Espectros. 249 N1,N5-Bis(4,6-dicloro-1,3,5-triazin-2-il)naftaleno-1,5-diamina (7): Espectro de IR de 7 (neto): 99,9 %T 99,75 796,60 781,17 1392,61 848,68 1597,06 1587,42 3234,62 99,45 1168,86 99,6 1504,48 99,3 1548,84 1238,30 99,15 99 3600 JR-23 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectros de UV, fluorescencia y excitación en CH2Cl2 de 7. UV Fluorescence Excitation 0.30 140 120 0.25 100 Absorbance 80 0.15 60 0.10 40 0.05 20 0.00 0 300 400 Wavelength (nm) 500 Intensity (a.u.) 0.20 250 4) 2,4-DIMETOXIBENCILAMINOTRIAZINAS: N-(2,4-Dimetoxibencil)-N´,N´´-bis-(2-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (9a). Espectro de 1H-RMN de 9a (DMSO, 80 ºC): N O N H N N N 9a H N N HN N Espectro de 1H-RMN de 9a diferentes temperaturas: N O Anexo 2. Espectros. 251 N2-(2,4-Dimetoxibencil)-N4,N6-di(naftalen-1-il)-1,3,5-triazina-2,4,6-triamina (9g). Espectro de 1H-RMN de 9g (DMSO, 60 ºC): HN N N NH N HN 9g OCH3 OCH 3 Espectros de 1H-RMN de 9g a diferentes temperaturas: 252 N-(2,4-Dimetoxibencil)-N´,N´´-bis-(3-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (9i). Espectro de 1H-RMN de 9i (DMSO, 85 ºC): O H N N N N H N N O HN N 9i N N Espectros de 1H-RMN de 9i a diferentes temperaturas: Anexo 2. Espectros. 253 N-(2,4-Dimetoxibencil)-N´,N´´-bis-(4-pirazol-1-ilfenil)-1,3,5-triazina-2,4,6-triamina (9j). Espectro de 1H-RMN de 9j (DMSO, 80 ºC): O H N N N N N H N N O HN 9j N N Espectros de 1H-RMN de 9j a diferentes temperaturas: 254 N-(2,4-Dimetoxibencil)-N´,N´´-bis-(1-fenilpirazol-4-il)-1,3,5-triazina-2,4,6-triamina (9k). Espectro de 1H-RMN de 9k (DMSO, 115 ºC). O N N H N N N N 9k N N Espectro de 1H-RMN de 9k a diferentes temperaturas: O Anexo 2. Espectros. 255 1) MONOAMINOTRIAZINAS: 4,6-Dicloro-1,3,5-triazina-2-amina (10) Espectro de IR de 10 (neto): 100 %T 99,5 99 846,75 98,5 97,5 796,60 1012,63 1251,80 1315,45 98 97 3219,19 96,5 3304,06 3387,00 95,5 1504,48 1643,35 96 95 94,5 94 4000 3600 MonoNH2triazina 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600 1/cm 256 N2,N4-Difenil-1,3,5-triazina-2,4,6-triamina (10b): Espectro de 1H-RMN de 10b (DMSO, 25 ºC): NH 2 N HN N N H N 10b Espectro de 13C-RMN de 10b (DMSO, 25 ºC): NH 2 N HN N N N H 10b Anexo 2. Espectros. 257 Espectro de IR de 10b (neto): 99 %T 754,17 3014,74 2968,45 97,5 1446,61 96 94,5 1363,67 91,5 1228,66 1217,08 93 90 1737,86 88,5 87 85,5 4000 I-34B 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 600 1/cm 258 N2,N4-Bis(4-metoxifenil)-1,3,5-triazina-2,4,6-triamina (10c): Espectro de 1H-RMN de 10c (DMSO, 25 ºC): NH 2 N HN O N N N H 10c O Espectro de 13C-RMN de 10c (DMSO, 25 ºC): NH 2 N HN O N N N H 10c O Anexo 2. Espectros. 259 Espectro de Espectro IR de 10c (neto): 97,5 817,82 1033,85 1020,34 1176,58 1489,05 1454,33 1433,11 1415,75 1506,41 1604,77 1575,84 1558,48 2968,45 90 2945,30 3014,74 3001,24 %T 82,5 1205,51 1354,03 75 1365,60 67,5 1228,66 1217,08 60 1737,86 52,5 45 3600 I-63Bh 3300 3000 2700 2400 2100 1950 1800 1650 1500 1350 1200 1050 900 750 1/cm 260 N2,N4-Bis(4-nitrofenil)-1,3,5-triazina-2,4,6-triamina (10h): Espectro de 1H-RMN de 10h (DMSO, 25 ºC). NH 2 N HN NO2 N N N H 10h NO2 Espectro de 13C-RMN de 10h (DMSO, 25 ºC). NH 2 N HN NO2 N N N H 10h NO2 Anexo 2. Espectros. 261 Espectro de IR de 10h (neto): 100 %T 1112,93 3014,74 2968,45 96 752,24 98 94 92 88 1217,08 86 1228,66 1363,67 90 1737,86 84 82 80 3600 I-35B 3300 3000 2700 2400 2100 1950 1800 1650 1500 1350 1200 1050 900 750 1/cm 262 2) DERIVADOS DE TRIAZINILGLICINA. Ácido 2-((4,6-bis((2-(1H-pirazol-1-il)fenil)amino)-1,3,5-triazin-2-il)amino)acético (13a): Espectro de 1H-RMN de 13a (DMSO, 25 ºC) en medio ácido: N N O H N N HO NH N N HN 13a N N Espectro de 13C-RMN de 13a (DMSO, 80 ºC) en medio ácido: N N O H N HO N N N HN 13a N NH N Anexo 2. Espectros. 263 Espectro de 1H-RMN de 13a (DMSO, 25 ºC) en medio básico: N N O H N O N N NH N HN N N Espectro de 13C-RMN de 13a (DMSO, 25 ºC) en medio básico: N N O O H N N N N HN N NH N 264 Espectros de 1H-RMN de 13a a diferentes temperaturas. Espectro de IR de la sal sódica de 13a (neto): 100 %T 796,60 810,10 99 1570,06 738,74 1591,27 97 939,33 1047,35 1614,42 3331,07 1327,03 1303,88 98 96 94 1408,04 1444,68 1506,41 95 93 3600 3200 JR-35 básico 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Anexo 2. Espectros. 265 Espectro IR de 13a (neto): 99 808,17 %T 1620,21 1328,95 96 941,26 1714,72 1047,35 1224,80 3309,85 97,5 1406,11 1556,55 94,5 752,24 93 1506,41 1494,83 91,5 1454,33 90 88,5 4000 JR-35 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 Espectro de UV, fluorescencia y excitación de 13a (CH2Cl2, 10-5 M): 0,05 8 UV Fluorescence Excitation 0,04 0,03 4 0,02 2 0,01 0,00 300 400 Wavelength (nm) 500 0 600 Intensity (a.u.) Absorbance 6 800 600 1/cm 266 Ácido 2-(4,6-bis(fenilamino)-1,3,5-triazin-2-il)amino)acético (13b): Espectro de 1H-RMN de 13b (DMSO, 80 ºC): O NH O N N H N N NH O 13b OH Espectro de 13C-RMN de 13b (DMSO, 80 ºC): O NH O N N H N N 13b NH O OH Anexo 2. Espectros. 267 Espectro de IR de 13b (neto): 100 %T 99,75 1024,20 99,5 686,66 1244,09 99,25 752,24 99 1666,50 1631,78 1608,63 98,5 1415,75 1402,25 1373,32 3292,49 98,75 1494,83 98,25 1444,68 98 97,75 3600 JR-15 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectro IR de 13b con ZnCl2 (neto): 100 %T 2924,09 879,54 1087,85 1415,75 1734,01 96 3421,72 98 754,17 1174,65 1174,65 1282,66 92 688,59 1450,47 1595,13 1556,55 1631,78 94 90 632,65 1026,13 1224,80 88 86 84 3600 3300 JR-15 Zn 3000 2700 2400 2100 1950 1800 1650 1500 1350 1200 1050 900 750 600 1/cm 268 Ácido 2-((4,6-bis((4-metoxifenil)amino)-1,3,5-triazin-2-il)amino)acético (13c): Espectro de 1H-RMN de 13c (DMSO, 25 ºC): NH N N H N N NH O 13c OH Espectro de 13C-RMN de 13c (DMSO, 25 ºC): NH N N H N N NH O 13c OH Anexo 2. Espectros. 269 Espectro de IR de 13c (neto): 100 %T 2829,57 99,75 3290,56 1296,16 99,5 827,46 1411,89 1481,33 1031,92 1375,25 1664,57 1598,99 99 1174,65 99,25 1238,30 98,75 98,5 1508,33 98,25 98 3600 JR-13 puro 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm 270 Ácido 2-((4,6-di(piperidin-1-il)-1,3,5-triazin-2-il-amino)acético (13d): Espectro de 1H-RMN de 13d (DMSO, 25 ºC): N N N N N NH O 13d OH Espectro de 1H-RMN de 13d (DMSO, 80 ºC): N N N N N NH O 13d OH Anexo 2. Espectros. 271 Espectro de 13C-RMN de 13d (DMSO, 25 ºC): N N N N N NH O 13d OH Espectro de 13C-RMN de 13d (DMSO, 80 ºC): N N N N N NH O 13d OH 2000 1800 1600 1639,49 1531,48 1487,12 1450,47 1400 1384,89 1367,53 1284,59 1200 1234,44 1000 1022,27 991,41 954,76 852,54 800 771,53 729,09 1/cm 684,73 659,66 272 2400 1672,28 1597,06 Espectro de IR de 13d (neto): 2800 2858,51 99 3200 2937,59 %T 97,5 96 94,5 93 91,5 90 88,5 3600 3267,41 Anexo 2. Espectros. 273 Ácido 2-((4,6-dimorfolino-1,3,5-triazin-2-il)amino)acético (13e): Espectro de 1H-RMN de 13e (DMSO, 80 ºC): O N N N N N NH O O 13e OH Espectro de 13C-RMN de 13e (DMSO, 80 ºC):: O N N N N N O NH O 13e OH 2000 1800 1600 1602,85 1556,55 1525,69 1489,05 1400 1394,53 1361,74 1301,95 1273,02 1257,59 1200 1238,30 1114,86 1068,56 1000 1029,99 1004,91 912,33 862,18 800 794,67 771,53 274 2400 1641,42 Espectro de IR de 13e (neto): 2800 1672,28 100 3200 2966,52 2914,44 2862,36 %T 99 98 97 96 95 94 93 3600 3296,35 1/cm Anexo 2. Espectros. 275 Ácido 2-(4,6-bis(difenilamino)-1,3,5-triazin-2-il)amino)acético (13f): Espectro de 1H-RMN de 13f (DMSO, 80 ºC): O H N HO N N N N N 13f Espectro de 13C-RMN de 13f (DMSO, 80 ºC): O HO H N N N N N N 13f 276 Espectro de IR de 13f (neto): 100 %T 1024,20 1215,15 1294,24 98 1649,14 1732,08 1714,72 99 748,38 1489,05 1463,97 1573,91 3417,86 96 806,25 97 690,52 95 1548,84 1537,27 94 1384,89 93 92 3600 JR-37 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 1/cm Espectro de UV, fluorescencia y excitación de 13f (CH2Cl2, 10-5 M). 6 0,025 UV Fluorescence Excitation 0,020 0,010 2 0,005 0 0,000 300 400 Wavelength (nm) 500 Intensity (a.u.) Absorbance 4 0,015 Anexo 2. Espectros. 277 Ácido 2-((4,6-bis(naftalen-1ilamino)-1,3,5-triazin-2-il)amino)acético (13g): Espectro de 1H-RMN de 13g (DMSO, 80 ºC): HO O NH N N N H N H N 13g Espectro de 13C-RMN de 13g (DMSO, 80 ºC): HO O NH N N H N N 13g N H 278 Espectro de IR de 13g (neto): 99,75 %T 3049,46 99 1392,61 779,24 759,95 1633,71 96 1589,34 1705,07 1508,33 96,75 673,16 3396,64 97,5 1244,09 3348,42 98,25 95,25 1556,55 94,5 93,75 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 Espectros de UV, fluorescencia y excitación de 13g (CH2Cl2, 10-5 M). 0,06 UV Fluorescence Excitation 0,05 12 10 8 0,03 6 0,02 4 0,01 2 0,00 0 300 400 Wavelength (nm) 500 Intensity (a.u.) Absorbance 0,04 14 600 1/cm Anexo 2. Espectros. 279 2-((4,6-Bis(naftalen-1ilamino)-1,3,5-triazin-2-il)amino)acetato de metilo (14g): Espectro de 1H-RMN de 14g (DMSO, 80 ºC): O HN N HN N N O NH 14g Espectro de 13C-RMN de 14g (DMSO, 80 ºC): O HN N HN N N 14g O NH 280 Espectro de IR de 14g (neto): 100 %T 99 1267,23 1394,53 97 96 1215,15 1747,51 98 1614,42 94 1496,76 1631,78 771,53 95 1556,55 93 92 3600 JR-42 3300 3000 2700 2400 2100 1950 1800 1650 1500 1350 1200 1050 900 750 1/cm 281 ANEXO 3. TABLAS ÚTILES Tabla A3.1. Distancias (Å) y ángulos (º) seleccionados para el compuesto 13a. O1-C5 O2-C5 N1-C1 N1-C2 N2-C3 N2-C2 N3-C1 N3-C3 N4-C1 N4-C4 N5-C2 N5-C11 N8-C3 N8-C2 C1-N1-C2 C3-N2-C2 C1-N3-C3 C1-N4-C4 C2-N5-C11 C3-N8-C21 1.329(5) 1.187(5) 1.297(5) 1.343(5) 1.311(6) 1.337(5) 1.344(6) 1.373(6) 1.327(6) 1.429(6) 1.332(6) 1.370(6) 1.315(5) 1.418(6) 114.4(4) 116.2(4) 119.1(5) 121.4(5) 134.9(5) 129.8(4) Tabla A3.2. Distancias (Å) y ángulos (º) seleccionados para el compuesto 13d. O1-C15 1.248(4) O2-C15 1.231(4) N1-C2 1.317(4) N1-C1 1.357(4) N2-C2 1.366(3) N2-C3 1.381(4) N3-C3 1.306(4) N3-C1 1.361(3) N6-C3 1.329(3) N6-C14 1.449(4) C14-C15 1.532(4) 282 C2-N1-C1 117.1(2) C2-N2-C3 117.4(3) C3-N3-C1 115.2(2) C3-N6-C14 122.2(3) O2-C15-O1 127.4(3) O2-C15-C14 115.3(3) O1-C15-C14 117.3(2) Tabla A3.3. Datos obtenidos para calcular el límite de detección de [Hg(II)]. Medida 1 15,26 15,72 33,58 50,51 68,78 82 91,75 97,45 101,67 Medida 2 15,38 15,73 33,84 50,27 68,59 81,66 91,77 97,62 101,58 Medida 3 15,35 15,77 33,85 50,62 68,32 81,56 92,11 97,56 101,74 I media 15,33 15,74 33,76 50,47 68,56 81,74 91,88 97,54 101,66 Log (I) 1,186 1,197 1,528 1,703 1,836 1,912 1,963 1,989 2,007 Log [Hg] -9 -8,9208 -8,8538 -8,7958 -8,7447 -8,6989 -8,6575 -8,6382 -8,5228 2+ Conc. Hg 1,00E-09 1,20E-09 1,40E-09 1,60E-09 1,80E-09 2,00E-09 2,20E-09 2,30E-09 3,00E-09