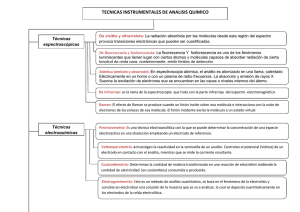
Guia sobre figuras de mérito
Anuncio

Análisis Instrumental Métodos de estimación de Límite de Detección y Límite de Cuantificación en metodologías instrumentales 1. Algunas definiciones El proceso de validación de una metodología analítica, obligatorio en determinados rubros de actividad, incluye la determinación de su Límite de Detección y su Límite de Cuantificación. Sin embargo, los diferentes organismos internacionales los han definido de distinta forma y basándose en diferentes conceptos. Límite de detección El límite de detección, expresado como una concentración (LD), se calcula a partir de la medición más pequeña (yL), que puede ser detectada con certidumbre razonable en un determinado procedimiento analítico. (IUPAC). El concepto de exactitud se ha modificado en los últimos veinte años. La definición clásica era “exactitud de una medición es la coincidencia entre el resultado de una medición y el valor verdadero del mensurando” (ISO, Metrología, 1993). En la actualidad se estima la incertidumbre de una metodología como la contribución de las incertidumbres sistemáticas (exactitud) y los incertidumbres aleatorias (precisión). Repetitividad de una metodología Es la precisión de los resultados obtenidos a partir de determinaciones independientes del mismo mensurando, con el mismo método, por el mismo operador utilizando el mismo equipamiento y en el mismo laboratorio, y en un intervalo corto de tiempo (ISO 3534-1). El concepto de determinaciones independientes significa que se realiza la totalidad de la metodología analítica en cada replicado. Si no fuera así se estaría evaluando únicamente la “repetitividad instrumental” Reproducibilidad de una metodología Es la precisión de los resultados que se obtienen de determinaciones independientes del mismo mensurando, modificando uno de los parámetros que se definen para determinar la repetitividad. Como se muestra en la tabla 1. Tabla 1.- Comparación entre repetitividad y reproducibilidad. Repetibilidad Reproducibilidad Intra-laboratorio Reproducibilidad intermedia Ensayos de colaboración Muestra Misma Misma Misma Operador Mismo Diferente Tiempo Mismo Diferente Equipo Mismo Reactivos Mismos Diferentes Laboratorio Mismo Diferente Condiciones Se modifican una o mas condiciones, pero no todos Diferente Error de una medición Es una medida de la exactitud de un método, y se define como la diferencia que existe entre el resultado de una medición y el “valor verdadero”. Pero, debido a que no es posible conocer el “valor verdadero” de la concentración de un analito, tampoco es posible conocer el error cometido en una determinación. Sin embargo, podemos asumir como valor verdadero al obtenido a partir de una gran cantidad de análisis de la muestra en distintos laboratorios. Es asi como organismos como el NIST comercializan materiales de referencia certificados (CRM) para poder testear la exactitud de los métodos analíticos. Incertidumbre de una medición Es el parámetro asociado con el resultado de una medición, que caracteriza la dispersión de los valores que pueden atribuirse razonablemente al mensurando. Se trata de un intervalo estimado, que se informa junto al resultado calculado, y en el que existe una cierta probabilidad (usualmente 95 - 99%) de encontrar al valor verdadero. A diferencia del error, la incertidumbre nunca puede ser 0, y si bien incluye la repetitividad es obviamente mayor que ella. Como se observa en la figura 1. Figura 1.- descripción gráfica de las definiciones de error incertidumbre e intervalo de confianza 2. Límite de detección basado en la repetitividad del blanco Consideraciones conceptuales Nunca se debe olvidar que las matemáticas asociadas a la medición son un auxiliar más de la misma, pero no constituyen el objeto central. De esta forma, cuando se encuentran discrepancias entre el resultado de aplicar una determinada estadística y la evidencia química, deberá tener prioridad esta última y en todo caso encontrar las razones de la discrepancia. Habitualmente el mal uso de fórmulas o el no cumplimiento de condiciones de validez imprescindibles para determinado modelo estadístico sugieren valores de cifras de mérito muy alejadas de la realidad. Por ejemplo, si contamos con un grupo de muestras de las que algunas contienen cierto analito y otras no, y mediante un determinado método somos capaces de determinar cuales son las que lo contienen, es claro que estamos por sobre el límite de detección. Esto es particularmente cierto cuando se trata de un número de muestras tal que la probabilidad estadística de haber cometido errores repetidas veces se hace despreciable. Sin embargo, la aplicacion cruda de la estimación del LD mediante el análisis de una curva de calibración puede dar valores tan altos que indiquen que es imposible medir en esas condiciones. Esto suele deberse a la incertidumbre acumulada en la preparacion de patrones, que puede ser muy alta, mientras que el “valor cero” del analito es muy probable que tenga una incertidumbre mucho menor, sobre todo en matrices sencillas. Definición de LD considerando solo error de tipo I La primera aproximación definió límite de detección como: “La concentración de analito que origina una señal que puede diferenciarse estadísticamente del blanco”. Si se asume que la señal del blanco tiene distribución gaussiana, entonces la probabilidad de que la más pequeña señal analítica discernible, yL, puede ser medida y no corresponde a una fluctuación aleatoria de la medida del blanco, depende de a cuantas unidades (k) de desviación estándar del blanco (sB) está yL de yB (promedio de las medidas del blanco). Si la diferencia entre yB e yL es 3sB la probabilidad que la medida de yL sea una fluctuación del blanco es menor que el 0,13% (si el número de medidas del blanco es superior a 25). A esto se lo conoce como la probabilidad de falso positivo o error de tipo I (Ver figura 2). Figura 2.- Distribución de probabilidad de las medidas del blanco. Entonces la señal mínima que se diferencia estadísticamente de la blanco (yL) se puede calcular y L y B 3s B [1] Luego el valor de concentración teórico que debería tener tal señal surge de reemplazar la ecuación [1] en la recta de calibración (y = mX + yB) dando el tradicional cálculo del LD LD 3s B m [2] El límite de cuantificación se define como la concentración de analito que origina una señal que puede cuantificarse con una precisión acotada (usualmente 10%). Por lo tanto, aproximando que la precisión es constante en el rango inferior de la curva de calibración se puede determinar el límite de cuantificación (LC) como: LC 10s B m [3] Este método utiliza como fuente de variabilidad de las señales, la desviación estándar del blanco. Para conocerlo con precisión es necesario repetir la medición del blanco un gran número de veces (n>20), lo que muchas veces resulta muy costoso y consume tiempo. Límite de detección basado en la curva de calibración Como estimadores alternativos al sB se suelen utilizar: La desviación estándar de los residuos del calibrado (sy/x, ecuación 4), el desvío estándar de la ordenada al origen (sb, ecuación 5), y la extrapolación de las hipérbolas de confianza al eje y (s0, ecuación 6). (Note que en el último caso no es necesario dividir por la pendiente para el cálculo del LD). Esto es útil si no se ha repetido el blanco y s B no puede calcularse. n (y sy/x = i 1 i yˆ i ) 2 [4] n2 sb = s y / x ; 1 n x2 m (x i 1 s0 = sy/ x m 1 1 p n x2 m (x i 1 i x) i x) [5] 2 [6] 2 Estas aproximaciones son válidas cuando la precisión instrumental es constante a lo largo del ámbito de linealidad, y la variabilidad asociada a la preparación de los estándares de calibración es despreciable frente a la variabilidad instrumental. Si esto no se cumple, el uso de estos parámetros en el cálculo producirá una sobreestimación del LD. Hay que tener en cuenta que con los instrumentos actuales es muy probable que la variabilidad de preparacion de estandares sea similar o mayor a la variabilidad instrumental. Definición de LD considerando error de tipo I y II ¿Qué ocurre si se mide una solución de analito de concentración igual al límite de detección? Como la distribución de probabilidad será una gaussiana centrada en la mínima señal detectable (yL), la mitad de las veces tendrá un valor mayor a yL y la otra mitad será menor. Con lo cual la mitad de las veces no se detectará, y la tasa de falso negativo será del 50%. Esto quiere decir que la mitad de las veces que se mide una concentración de analito igual a su límite de detección no será detectable. (ver figura 3). Figura 3.- Distribución de probabilidad de las medidas del blanco (curva azul), y distribución de probabilidad de las medidas de una solución de analito de concentración igual al LD (curva roja). Las áreas sombreadas representa la probabilidad de cometer error de tipo I (area azul, ) y error de tipo II (área roja,). Definición moderna del Límite de detección de IUPAC Para solucionar este inconveniente IUPAC reformulo la definición de límite de detección con una sutil diferencia: “Es la menor concentración de analito que puede ser detectada con un cierto nivel de confianza”. Esta definición no solo limita el error tipo I que se puede cometer sino que también acota el error de tipo II. Entonces, bajo la nueva definición, el límite de detección ya no es el valor a partir del cual se toma la decisión de detectado, y pueden detectarse concentraciones de analito aun menores que el LD. Además se agregó la definición de un nuevo parámetro denominado como nivel crítico (Nc), que representa el Nivel de concentración límite a partir del cual se toma la decisión de “detección” del analito. En la definición moderna, el límite de detección (LD) se calcula en función del desvío estándar de la concentración predicha para una muestra blanco (s0). Para estimar s0, en lugar de repetir la medición del blanco un gran número de veces, IUPAC recomienda recurrir a la ecuación [6]. Como se muestra en la Figura 3, el LD se calcula mediante una prueba de hipótesis estadística. En primer lugar se fija la concentración llamada nivel crítico (NC en la Figura 4), a partir de la cual se toman decisiones respecto de la detección del analito. Para concentraciones superiores a NC, existe una probabilidad de cometer el llamado error de tipo I o falso positivo. Este último consiste en aceptar erróneamente la hipótesis alternativa, admitiendo que el analito está presente cuando en realidad está ausente. Como se aprecia en la Figura 1, la probabilidad de cometer este error de tipo I está dada por la zona sombreada de azul (área ), siendo la "distancia" de LC al cero de la escala igual al producto de s0 por el coeficiente t. Si se toma igual a 0,05, entonces una concentración superior a LC tendrá sólo un 5% de probabilidad de constituir un falso positivo. Figura 4.- Representación gráfica de la definicion moderna IUPAC para estimar el límite de detección. NC es el nivel crítico, LD el límite de detección, y las probabilidades correspondientes a errores de tipo I y II respectivamente, s0 el desvío estándar del blanco (en unidades de concentración) y t y t los coeficientes de student para grados de libertad. Del mismo modo, existe una probabilidad de cometer un error de tipo II o falso negativo, en el que se acepta erróneamente la hipótesis nula, admitiendo que el analito está ausente cuando en realidad está presente (zona sombreada de rojo en la Figura 1, con probabilidad igual a ). Si se toma también como 0,05, la probabilidad de obtener un falso negativo será del 5%. En este caso la distancia de LC a la concentración correspondiente a dicho valor de es el producto del coeficiente t por s0, considerando que este último parámetro es muy cercano al desvío estándar en la concentración de una muestra blanco. Puede notarse entonces que el valor de LOD depende de y , y de los desvíos estándar de las dos curvas gaussianas de la Figura 1. En general, ambas probabilidades se toman como iguales 0,05, mientras que los desvíos estándar se suponen ambos iguales a s0. De este modo, el LOD está dado por: LOD = 2 t0,05,m–2 s0 [7] Definición que ha sido adoptada también por IUPAC e ISO. En la práctica, dado que m es un número relativamente grande, el valor de (2t0,05,m–2) tiende a 3,28, por lo que una ecuación aproximada para el límite de detección es LD = 3,28 s0. Análogamente, el límite de cuantificación (LC) es la mínima concentración cuantificable en forma confiable. Este parámetro se toma como la concentración correspondiente a 10 veces el desvío estándar (en unidades de concentración) del blanco, con lo cual: LOQ = 10 s0 [8] De este modo, el desvío estándar relativo (DSR) para una concentración igual al LC es del orden del 10%, nivel que se toma convencionalmente como el máximo desvio estándar relativo aceptable para cuantificar el analito en una muestra. En la figura 5 se resumen las decisión tomadas a diferentes concentraciones. Figura 5.- Representación gráfica de las decisiones tomadas en función de la concentración 3. Métodos empíricos para la estimación del Límite de Cuantificación. Límite de cuantificación basado en la desviación standard relativa (RSD) de la señal. Este método considera la evidencia empírica de que la RSD de la señal aumenta cuando la concentración disminuye. El límite de cuantificación es la cantidad analítica para el cual la RSD de la señal neta del análisis alcanza un nivel preestablecido (0.1 ó 0.05). Se deben realizar análisis sucesivos a distintas concentraciones próximas al Límite de Cuantificación. Se grafica la RSD de la señal neta como una función de la concentración (gráfico logarítmico), entre el valor de LC esperado y 20-50 veces este valor. Se calcula la regresión lineal en este gráfico y el LC será la concentración para el RSD preestablecido (0,1 o 0,05) A modo de ejemplo Figura 6.- Desvio estándar relativo porcentual (%RSD) de la señal de Zn a 231nm obtenida por ICP-AES en función de su concentración usando una escala log-log. Resultan Y LC = 30 µ/l LC = 60 µ/l si RSD = 10% si RSD = 5% Límite de cuantificación del método o nivel mínimo El LC se deduce del límite de detección del método (MDL), definido como la concentración mínima de un analito que, en una matriz dada y con un método determinado, tiene una probabilidad del 99% de ser identificado, determinado cualitativamente, e informado como mayor que cero. Se deben realizar al menos 7 replicados de una adición de analito al blanco, con concentración entre 1 y 5 veces la esperada como límite de detección del método. Los replicados deben procesarse a través de todas las etapas analíticas del método. Considerando n replicados, se calcula la desvación standard s de las mediciones, y para n-1 grados de libertad y un nivel de confianza 1-α, resulta A partir del MDL, se calcula el nivel mínimo, que resulta ser el límite de concentración: Es de destacar que se trata de un límite muy realista, ya que considera la medición real de concentraciones próximas al LC. A diferencia del método anterior (de las desviaciones standard relativas), se está asumiendo que la desviación standard es constante frente a la concentración. Bibliografia 1. Alejandro C. Olivieri, Héctor C. Goicochea, La calibración en la Química Analitica, Ediciones UNL. 2. K. Danzer y L. A. Currie, Guidelines for calibration in analytical chemistry. Part 1. Fundamentals and single component calibration, Pure & Appl. Chem. 1998, 70, 9931014. 3. W. P. Gardiner, Statistical analysis methods for chemists. A software-based approach, The Royal Society of Chemistry, Cambridge, 1997. 4. J. N. Miller y J. C. Miller, Estadística y quimiometría para química analítica, 4ta. Edición, Prentice Hall, Madrid, 2002. 5. C. A. Clayton, J. W. Hines y P. D. Elkins, Detection limits with specified assurance probabilities, Anal. Chem. 1987, 59, 2506-2514. 6. L. A. Currie, Detection and quantification limits: origins and historical perspective, Anal. Chim. Acta 1999, 391, 127-134. 7. L. A. Currie, Recommendations in Evaluation of Analytical Methods including Detection and Quantification Capabilities, Pure Appl. Chem. 1995, 67, 1699-1723. 8. P. Wilrich, ISO/DIS 11843-1,2 (1995), Capability of Detection, ISO/TC69/SC6, ISO Standard, 11843-1, 1977. 9. M. Valcárcel, Principios de química analítica, Springer-Verlag Ibérica, Barcelona, 1999, p. 81. 10. Mermet, J.M. Spectrochimica Acta part B 63 (2008), 166- 182.