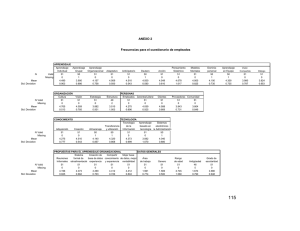
Máster en Estadística e Investigación Operativa
Anuncio

Máster en Estadística e Investigación Operativa Título: Inferencia estadística para el equilibrio de Hardy-Weinberg en estudios de genotipado con datos faltantes. Autor: Lic. Milagros Sánchez Mayor Director: Dr. Jan Graffelman Departamento: Departamento de Estadística e Investigación Operativa. Universidad: Universitat Politècnica de Catalunya Convocatoria: 2012 Índice general Índice general I 1 Introducción 1.1. Objetivos del trabajo . . . . . . . . . . . . . . . . . . . . . . . . . . . 1.2. Estructura del estudio . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 6 6 2 Conceptos básicos de la genética 2.1. ¿Qué son los SNPs? . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.2. La genética Mendeliana . . . . . . . . . . . . . . . . . . . . . . . . . . 2.3. Principio de Hardy-Weinberg . . . . . . . . . . . . . . . . . . . . . . . 2.3.1. Posibles desviaciones del Equilibrio Hardy-Weinberg . . . . . . 2.4. Pruebas estadı́sticas para las Proporciones de Hardy-Weinberg . . . . . 2.4.1. La prueba χ2 de Pearson . . . . . . . . . . . . . . . . . . . . . 2.4.2. Test exacto de Levene-Haldane . . . . . . . . . . . . . . . . . 2.4.3. Test de Razón de Verosimilitud (LRT) . . . . . . . . . . . . . . 2.5. Mı́nima frecuencia alélica (MAF) . . . . . . . . . . . . . . . . . . . . 2.6. Coeficiente de endogamia (f ) . . . . . . . . . . . . . . . . . . . . . . . 2.7. Potencia de las pruebas para detectar HWE . . . . . . . . . . . . . . . 2.7.1. Los cálculos de potencia de las pruebas clásicas para HWE . . . 2.7.2. Los cálculos de potencia de una prueba de HWE para la asociación marcadores-enfermedad . . . . . . . . . . . . . . . . . . . 2.8. Importancia y aplicación del Equilibrio de Hardy-Weinberg . . . . . . . 7 7 8 9 11 12 12 14 16 17 18 20 20 3 Descripción de la base de datos 3.1. Motivación por esta Base de Datos . . . . . . . . . . . . . . . . . . . . 3.2. Estructura de la base de datos . . . . . . . . . . . . . . . . . . . . . . . 23 23 24 4 Introducción a los Missing Data 4.1. Breve descripción del problema de los Missing Data en los SNPs . . . . 4.2. Missing Data en los SNPs . . . . . . . . . . . . . . . . . . . . . . . . . 27 27 27 I 21 22 ÍNDICE GENERAL II 4.3. Terminologı́as . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4.3.1. Mecanismos de Respuestas en Marcadores Genéticos . . . . 4.3.2. Patrones de Missing Data . . . . . . . . . . . . . . . . . . . 4.4. Teorı́a general de la imputación . . . . . . . . . . . . . . . . . . . . 4.4.1. Imputación Múltiple (IM) . . . . . . . . . . . . . . . . . . 4.4.2. Modelos de Imputación . . . . . . . . . . . . . . . . . . . . 4.4.3. Modelo de Localización General (GLM) . . . . . . . . . . 4.5. Análisis de sensibilidad . . . . . . . . . . . . . . . . . . . . . . . . 4.6. Metodologı́a de nuestro estudio de los Missing Data en el contexto HWE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 6 . . . . . . . . . . . . . . . . de . . 29 29 31 32 33 35 36 38 Análisis de los resultados 5.1. Estadı́stica Descriptiva de los SNPs completos . . . . . . . . . . . . . . 5.2. Inspeccionando los Missing Data . . . . . . . . . . . . . . . . . . . . . 5.2.1. Mecanismo de Patrones de Missing Data . . . . . . . . . . . . 5.3. Imputación Simple . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.4. Creando las imputaciones bajo MAR . . . . . . . . . . . . . . . . . . . 5.4.1. Creando las imputaciones con MICE . . . . . . . . . . . . . . 5.4.2. Selección de la matriz predictora . . . . . . . . . . . . . . . . . 5.4.3. Chequeando el diagnóstico de los Missing . . . . . . . . . . . . 5.4.4. Evidencia de sesgos en las imputaciones bajo MICE . . . . . . 5.4.5. Creando las imputaciones con CAT . . . . . . . . . . . . . . . 5.4.6. Chequeando el diagnóstico de los Missing . . . . . . . . . . . . 5.4.7. Evidencia de sesgo en las imputaciones bajo CAT . . . . . . . . 5.4.8. Creando las imputaciones con MIX . . . . . . . . . . . . . . . 5.4.9. Chequeando el diagnóstico de los Missing . . . . . . . . . . . . 5.4.10. Evidencia de sesgo en las imputaciones bajo MIX . . . . . . . . 5.4.11. Comparando las Imputaciones . . . . . . . . . . . . . . . . . . 5.5. Pooling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.6. Creando las imputaciones bajo MNAR. Análisis de sensibilidad . . . . 5.6.1. Chequeando el diagnóstico de los Missing . . . . . . . . . . . . 5.7. Comparación de modelos de imputación respecto a HWE . . . . . . . . 5.8. Número de Marcadores significativos bajo imputación . . . . . . . . . 5.9. Cálculo de la potencia y tamaño muestral . . . . . . . . . . . . . . . . 5.9.1. Potencia de las pruebas clásicas de HWE . . . . . . . . . . . . 5.9.2. Potencia de la prueba de HWE para la asociación marcadoresenfermedad . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 41 45 47 52 54 54 54 57 60 62 63 63 64 64 65 66 67 70 72 74 75 78 78 Discusión y conclusión 83 Bibliografı́a 38 80 87 ÍNDICE GENERAL III Índice de figuras 91 Índice de tablas 93 Dedicatoria A mis padres que son el centro de mi universo y a mi sobrino que es como un hijo... A Julian por entregarme tanto de ti y estar siempre presente... 1 Agradecimientos Agradezco a mis padres que son la razón de mi vida, a mi madre por todo su amor, a mi padre por ser mi guı́a. A mi tutor Dr. Jan Graffelman, no tengo palabras para expresarle todo mi agradecimiento y cariño que siento, porque ha sido incondicional conmigo. Siento mucho orgullo de haberlo conocido, porque es excelente profesional y excelente persona. Por haberme llevado de la mano en este largo viaje de 2 años y en nuestro trabajo, sin dejar que perdiera el rumbo. Porque más que un tutor ha sido un amigo y me ha sabido orientar en mis desconocimientos. Ha sido una de las personas que ha hecho que mi mentalidad cambiase respecto a este mundo fuera de mi paı́s. Gracias por todo. A mi gran amigo cubano, Deivy Wilson, por ser un ejemplar amigo y un gran cubano. A mis amigos españoles: Belchin Adriyanov Kostov, Nuria Planell, Sara Fisas, Susana Santiago, Xavier Puerta y Juan Carlos Martı́n, que siempre me tienen presente y han logrado que yo viese otros horizontes. A todos mis profesores del máster, en especial a los profesores Marta Pérez, Jan Graffelman, Tomás Aluja, Mónica Bécue y Eric Cobo, por la intensidad de sus clases que hacı́an que me sintiera muy bien en ellas. Al Dr. Victor Moreno que nos ha proporcionado los datos. A Samantha Cook (Post.PhD), por orientarnos en nuestros inicios de este trabajo. A mis amigos que están en Cuba, que dı́a a dı́a se comunican conmigo para darme ánimos y fuerzas para seguir este largo camino. A mi paı́s que me formó como profesional y que además extraño mucho. Gracias por todo a todos. 3 Capı́tulo 1 Introducción Las razones para que existan los Missing Data pueden ser diversas, particularizando en el ámbito de la genética el hecho de los Missing Data se asocian a causas como: problemas coligados a la calidad del marco del muestreo, fallos en los instrumentos de medida, pérdida de la muestra, los sujetos no asisten a la consulta (en diseños longitudinales pueden abandonar el estudio en un momento concreto). Otras causas son los errores informáticos, un ejemplo puede ser a la hora de entrar los datos a el software provocando pérdidas de los datos o desajustes de las variables, o cuando se concatenan bases de datos, etc. De aquı́ que la presencia de Missing Data es un problema común a cualquier investigación, que cada vez va en aumento y por tanto no puede ser ignorado en el estudio que se desea realizar. Ignorar los Missing Data puede tener o no repercusiones graves, en caso positivo estas repercusiones van desde la pérdida de potencia del estudio por la eliminación de observaciones, de variables y por tanto la reducción de la capacidad de detectar las relaciones reales de los datos, etc., hasta la aparición de sesgos inaceptables. La eliminación de sujetos y por ende la reducción del tamaño muestral, la imputación de valores sin criterio, etc., limita la validez interna y de ahı́ su representatividad o validez externa de los resultados del estudio. En nuestro trabajo nos basaremos en las variaciones genéticas y la repercusión de la presencia de Missing Data en ellos, donde la mayorı́a de estas variaciones humanas se ven influenciadas por los genes y estos se remontan a los SNPs, figura 1.1, es decir, a los polimorfismos de nucleótido simple (SNP). El SNP es el marcador genético más sencillo, que consiste en una variación en la secuencia 5 Figura 1.1: Comparación entre ADN’s 6 CAPÍTULO 1. INTRODUCCIÓN de ADN que afecta a una sola base (adenina (A), timina (T), citosina (C) o guanina (G)) de una posición en la secuencia del genoma. Los SNPs se producen una vez cada 300 nucleótidos en promedio, lo que significa que hay aproximadamente 10 millones de SNPs en el genoma humano. Pueden actuar como marcadores biológicos o comúnmente llamados marcadores genéticos para localizar aquellos genes que pueden afectar a la respuesta de los individuos a enfermedades, bacterias, virus, productos quı́micos, fármacos, vacunas, etc. Los SNPs también sirven para el análisis de los patrones de variación genética molecular para reconstruir la historia evolutiva de las poblaciones humanas, dicho de otra manera, los SNPs han pasado a ser uno de los marcadores más importantes de la investigación genética, especı́ficamente en la investigación biomédica, ya que proporcionan pistas para nuevos objetivos, principalmente en la comparación de regiones del genoma entre las cohortes, etc. 1.1. Objetivos del trabajo Los marcadores genéticos, los SNPs entre ellos, cumplen en general una ley básica, la ley de Hardy Weinberg (HWE) y existen varios procedimientos estadı́sticos para comprobar si marcadores genéticos concuerdan con esta ley o no. En las pruebas estadı́sticas para HWE siempre se descartan los genotipos faltantes. El objetivo de este trabajo consiste en llevar a cabo inferencia para HWE teniendo en cuenta los datos faltantes. 1.2. Estructura del estudio Hemos planteado un capı́tulo referente a los conceptos básicos de la genética (Capı́tulo 2), en él hemos descrito la mayorı́a de las técnicas de estadı́stica descriptivas comúnmente usadas para el tratamiento de los SNPs. El Capı́tulo 3 es concerniente a la base de datos referente a pacientes de Cáncer de Colon; aquı́ explicamos muy detalladamente la estructura de dicha base de datos, para qué se usó y con qué objetivo la tomamos, también hacemos una pequeña referencia de dónde se obtuvo. El capı́tulo siguiente (4) se dedica a los missing data y toda una teorı́a basada en ellos. El último capı́tulo (5) expone los resultados de los análisis. Al final se presentan las referencias en una bibliografı́a. Capı́tulo 2 Conceptos básicos de la genética 2.1. ¿Qué son los SNPs? Polimorfismos de nucleótido simple (SNP), son el tipo más común de variación genética entre las personas o dentro de una misma persona. Cada SNP representa una posición en la cadena del ADN que muestra variabilidad. [1] Los nucleótidos son moléculas orgánicas que están formados por 3 componentes fundamentales: 1. Bases nitrogenadas: ? Purı́nicas: Adenina (A) y la Guanina (G). Ambas forman parte del ADN y ARN. ? Pirimidı́nicas: Timina (T), Citocina (C) y el Uracilo (U). La Timina y Citocina intervienen en la formación del ADN. En el ARN aparecen la Citocina y el Uracilo. ? Isoaloxacı́nicas: Flavina (F). No forma parte del ADN ni ARN. 2. Pentosa: Es el azúcar de 5 átomos de carbono. 3. Ácido Fosfórico: Cada nucleótido puede contener de 1 a 3 grupos fosfato. Por lo tanto los SNPs son variaciones en la secuencia del ADN, que esta ocurre cuando una de las bases nitrogenadas del nucleótido que intervienen en el ADN (A, G, T, C) es alterada y por ende es alterada la secuencia del Genoma. [1] 7 8 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA Estas diferencias pueden existir entre miembros de una misma especie, [2] como se puede ver en la figura 2.1 Figura 2.1: 2 SNPs mostrando variabilidad entre individuos de una misma especie Las diferencias también pueden manifestarse en pares de cromosomas en un mismo individuo, es decir, si una posición muestra variabilidad, también será posible observar variabilidad dentro de un mismo individuo, si se trata de un heterocigoto, figura 2.2 (concepto que explicaremos más adelante): Figura 2.2: Un individuo que es heterocigoto para un determinado SNP El ser humano tiene un total de 23 pares de cromosomas por lo que hablamos de individuos diploides, 22 pares que son autosomas y 1 par referente al sexo. En total hay 46 cromosomas. Los organismos diploides tienen 2 copias de cada gen, una ubicado en el cromosoma recibido de la madre y otro en el cromosoma recibido del padre. 2.2. La genética Mendeliana En la práctica casi todos los SNPs son bi-alélicos, esto significa que para 2 fragmentos de una secuencia de ADN en diferentes individuos, por ejemplo: AAGCCTA - 9 2.3. PRINCIPIO DE HARDY-WEINBERG AAGCTTA, contiene una diferencia en un nucleótido simple, en estos casos decimos que hay 2 alelos: C y T. Una variación alélica en una posición (locus) se manifiesta al nivel de individuos por la existencia de 3 tipos de individuos, es decir, por ejemplo para 2 alelos: A y T, tenemos 3 tipos de combinaciones en individuos diploides: √ 2 Alelos: AA (Homocigótico para el alelo A) √ 2 Alelos: TT (Homocigótico para el alelo T) √ Un Alelo A y un Alelo T: AT (Heterocigótico A y T) Tipos de Alelos Los alelos describen las diversas formas que adopta un gen detectado como diferentes fenotipos, que estas formas difieren en secuencia o función. • Los alelos que varı́an en secuencia tienen diferencias en el ADN debido a deleciones, inserciones o sustituciones. En general lo más común son las sustituciones. • Los alelos que difieren en función pueden o no tener diferencias conocidas en las secuencias, pero se evalúan por la forma en que afectan al organismo. En función de su expresión en el fenotipo se pueden dividir en: • Alelos dominantes: aquellos que aparecen en el fenotipo de los individuos heterocigotos o hı́bridos para un determinado carácter, además de en el homocigoto. • Alelos recesivos: los que quedan enmascarados del fenotipo de un individuo heterocigoto y sólo aparecen en el homocigoto, siendo homocigótico para los genes recesivos. 2.3. Principio de Hardy-Weinberg El principio de hardy-Weinberg fue formulado en el 1908 independientemente por Godfrey Harold Hardy un eminente matemático inglés y por Wilhelm Weinberg un médico alemán (figura 2.3); Hardy conocido por sus logros en la teorı́a de números y el análisis matemático y Weinberg un médico gineco-obstetra que ejercı́a en Stuttgart. Figura 2.3: Hardy (arriba) Weinberg (abajo) 10 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA El principio de Hardy-Weinberg está basado en las frecuencias alélicas y genotı́picas de una población donde este se define en que estas frecuencias se mantengan constantes de generación en generación, es decir, se encuentren en equilibrio a menos que se introduzcan influencias perturbadoras [3]. Este equilibrio genético es un estado ideal que ofrece una lı́nea para medir el cambio entre generaciones, por eso se dice que es imposible en la naturaleza. Con la ley de Hardy-Weinberg se asentaron los cimientos de la genética de poblaciones, según la cual, la alteración genética de una población sólo puede darse por factores como mutaciones, selección natural, influencias casuales, convergencias o divergencias individuales, de modo que el cambio genético implica la perturbación del equilibrio establecido por la ley de Hardy-Weinberg, que seguidamente explicaremos. El Principio de Hardy-Weinberg relaciona las frecuencias alélicas con las frecuencias genotı́picas en una población de individuos diploides o poliploides. Caso diploide: El caso diploide es el caso más simple. Denominemos un alelo como A y otro como T y sean p y q sus frecuencias respectivamente. Bajo la condición p + q = 1 tendrı́amos, si la población está en equilibrio, la siguiente tabla: Figura 2.4: frecuencias de Hardy-Weinberg Donde p2 es la frecuencia para AA (homocigotos), q 2 es la frecuencia para TT (homocigotos) y 2pq es la frecuencia para AT (heterocigotos). Se alcanza el equilibrio en una sola generación de apareamiento aleatorio. Estas frecuencias son las llamadas frecuencias de Hardy-Weinberg o Proporciones de Hardy-Weinberg. Como habı́amos comentado, podemos bajo la condición p + q = 1 expresar las frecuencias genotı́picas como la expansión binomial (p + q)2 = 1 ⇐⇒ p2 + 2pq + q 2 = 1 (2.1) Generalización para el caso de más de 2 alelos: Consideraremos un alelo extra, con frecuencia r, entonces la expansión trinomial serı́a: (p + q + r)2 = 1 ⇐⇒ p2 + q 2 + r2 + 2pq + 2pr + 2qr = 1 (2.2) Sucesivamente podemos extenderlo a n alelos, es decir, sean A1 , . . . , An alelos y sus respectivas frecuencias alélicas p1 , . . . , pn 2.3. PRINCIPIO DE HARDY-WEINBERG 11 Para el caso de alelos múltiples en un locus diploide tenemos las proporciones Hardy-Weinberg siguientes: F rec(Ai Ai ) = p2i =⇒ dado para homocigotos F rec(Ai Aj ) = 2pi pj =⇒ dado para heterocigotos (2.3) El número de posibles genotipos G con un número de alelos n está dado por la expresión: G = [n(n + 1)]/2 Generalización para poliploide: El caso poliploide consta cuando un organismo tiene más de 2 copias de cada cromosoma, para la cual se cumple también el Equilibrio de Hardy-Weinberg. Sean c el número de ploidı́a por lo tanto para el caso poliploide tenemos la expansión polinomial (p + q)c Generalización completa: Sean n alelos en c-ploidı́a, las frecuencias genotı́picas en el Equilibrio de Hardy-Weinberg están dadas por la expansión multinomial de: (pi +. . . +pn )c X c (pi + . . . + pn )c = (2.4) pk1 · · · pk1n k1 , . . . , kn 1 k1 ,...,kn ∈ℵ:k1 +...+kn =c 2.3.1. Posibles desviaciones del Equilibrio Hardy-Weinberg La ley de Hardy-Weinberg se basa en una serie de supuestos que enumeramos a continuación [3]: 1. Apareamiento Aleatorizado: Cuando no ocurre el apareamiento aleatorizado las proporciones de Hardy-Weinberg no existen y estás sólo estarán dadas en las frecuencias de los genotipos después de una generación de apareamiento aleatorizado dentro de la población. El apareamiento no aleatorizado puede ocurrir de 3 maneras: a. Endogamia: La que provoca un aumento de la homocigosidad para todos los genes. b. Apareamiento selectivo: Provocando un aumento de la homocigosidad sólo para los genes implicados en el rasgo que es selectivamente acoplado. c. Población de tamaño pequeño: Conlleva un cambio aleatorio en las frecuencias genotı́picas. Llamado Desvı́o Genético. Las demás suposiciones afectan a las frecuencias alélicas, pero no afectan por sı́ mismas al apareamiento aleatorio. Si una población viola alguna de estas, la población seguirá teniendo proporciones de Hardy-Weinberg en cada generación, pero las frecuencias alélicas cambiarán con esa fuerza. 12 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA 2. Ausencia de Selección Natural: Causa un cambio en las frecuencias alélicas muy rápidamente, mientras que la selección direccional al final conduce a la pérdida de todos los alelos excepto el favorecido. Hay dos tipos de selección: a. Selección de Mortalidad: Ciertos genotipos son menos eficaces que otras para sobrevivir hasta el final de su periodo reproductivo. Selección de Mortalidad es simplemente otra manera del criterio de aptitud de Darwin: La Supervivencia. b. Selección de Fecundidad: Ciertos fenotipos (por lo tanto genotipos) pueden hacer una contribución desproporcionada de la siguiente generación, es decir, un número desproporcionado de jóvenes en la próxima generación. La Selección de Fecundidad es otra forma del criterio de aptitud de Darwin: Tamaño de la Familia. 3. Ausencia de Mutación: Este tendrá un efecto muy sutil en las frecuencias alélicas. Las tasas de mutación son del orden de 10−4 a 10−8 por locus por generación. La Mutación recurrente mantendrá los alelos en la población, incluso si hay una fuerte selección en contra ellos. 4. Ausencia de Migración: Genéticamente une 2 o más poblaciones en conjunto. En general las frecuencias alélicas se harán más homogéneas entre las poblaciones. Algunos modelos de migración incluyen inherentemente el apareamiento no aleatorio, para estos modelos las proporciones de Hardy-Weinberg no suelen ser válidos. 5. Ausencia de Flujo de Genes: Es simplemente el flujo de genes entre las especies en lugar de dentro de una misma especie. Esta desviación aumenta la variabilidad de los genes, mediante la hibridación, introgresión, etc. 6. No hay Errores de genotipado: Confusión entre homocigotos y heterocigotos a la hora de la clasificación del genotipo. 2.4. 2.4.1. Pruebas estadı́sticas para las Proporciones de HardyWeinberg La prueba χ2 de Pearson La comprobación de la desviación de las Proporciones de Hardy-Weinberg (PHW) se suele llevar a cabo utilizando la prueba χ2 de Pearson [4], mediante las frecuencias genotı́picas observadas que se han obtenido de los datos y las frecuencias genotı́picas esperadas bajo equilibrio [5]. El planteamiento de la hipótesis nula, es que en la población existen las proporciones de Hardy-Weinberg y la alternativa es que no existen las proporciones de HardyWeinberg en la población [2]. Definamos primero los números observados de genotipos [5]. 2.4. PRUEBAS ESTADÍSTICAS PARA LAS PROPORCIONES DE HARDY-WEINBERG 13 n = nAA + nAT + nT T o nAA = N Observado de Homocigotos AA = nAT = No Observado de Heterocigotos AT n = No Observado de Homocigotos TT TT (2.5) El procedimiento a seguir serı́a: 1. Calcular las frecuencias alélicas. 2 nAA + nAT 2 [nAA + nAT + nT T ] 2 nAA + nAT q =1−p=1− 2 [nAA + nAT + nT T ] 2 nT T − nAT = 2 [nAA + nAT + nT T ] p= (2.6) 2. Calcular los valores esperados de Hardy-Weinberg: E [AA] = np2 E [AT ] = 2npq (2.7) E [T T ] = nq 2 3. Calcular los grados de libertad (gl): NG: Número de Genotipos NA: Números de Alelos gl=NG-NA 4. Por lo tanto la prueba χ2 de Pearson serı́a: X2 = X AA,AT,T T (Obs - Esp)2 Esp (2.8) Donde para el caso de los SNPs bialélicos X 2 sigue una distribución χ2 con un grado de libertad y para los sistemas en los que hay un gran número de alelos, esto puede ofrecer datos con muchos genotipos de frecuencias cero y poca cantidad de genotipos, porque a menudo no hay suficientes individuos en la muestra para representar adecuadamente todas las clases genotı́picas. Si este es el caso, entonces la suposición asintótica de la distribución χ2 no se sostendrá y puede ser necesario utilizar el test exacto de LeveneHaldane. Veamos el procedimiento descrito mediante un ejemplo. 14 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA 1. Calcular las frecuencias alélicas. 2. Calculamos los valores esperados: E [AA] = 94 · 0,6063832 = 34,56383 E [AT ] = 2 · 94 · 0,606383 · 0,393617 = 44,87234 E [T T ] = 94 · 0,3936172 = 14,56383 3. Calcular los grados de libertad (gl): NG = 3 tipos de Genotipos NA = 2 Alelos gl = 1 4. Por lo tanto la prueba χ2 de Pearson serı́a: (38 − 34,56383)21 (38 − 44,87234)2 (18 − 14,56383)2 + + = 2,2049 X = 34,56383 44,87234 14,56383 2 El nivel de significancia del 5 % para un grado de libertad es de 3.84, como el valor obtenido es menor, implica que no podemos rechazar la hipótesis nula de que la muestra indicada está en equilibrio de Hardy-Weinberg. El valor p de la prueba es P (χ21 ≥ 2,2049) = 0,1376 2.4.2. Test exacto de Levene-Haldane El test exacto se puede aplicar para comprobar si existen proporciones de HardyWeinberg. Como el test está condicionado por las frecuencias alélicas, p y q, el problema se puede entender como la comprobación del número adecuado de heterocigotos. De esta forma, la hipótesis de las proporciones de Hardy-Weinberg queda violada si el número de heterocigotos es muy grande o muy pequeño. Las probabilidades condicionadas para el heterocigoto, dadas las frecuencias alélicas, las proporciona Emigh [4] de la forma: n h i nAA , nAT , nT T nAT Prob nAT nA = 2 (2.9) 2n nA 2.4. PRUEBAS ESTADÍSTICAS PARA LAS PROPORCIONES DE HARDY-WEINBERG 15 Donde nAA , nAT , nT T son los números observados para los 3 fenotipos AA, AT, TT y nA es el número de alelos A, cuyas expresiones son nA = 2nAA + nAT y nT = 2n − nA , además para una muestra, el máximo número de heterocigotos está dada por la expresión min(nA , nT ). Si realizamos el test para el ejemplo anterior obtenemos para los heterocigotos observados posibles sus probabilidades exactas en la siguiente tabla. No Heterocigotos 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 Nivel Significancia 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 0.0002 0.0009 0.0032 0.0099 0.0253 No Heterocigotos 38 40 42 44 46 48 50 52 54 56 58 60 62 64 66 68 70 72 74 Nivel Significancia 0.0533 0.0934 0.1365 0.1662 0.1686 0.1423 0.0997 0.0577 0.0275 0.0107 0.0034 0.0009 0.0002 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000 Haciendo los cálculos pertinentes para este ejemplo tenemos: nA = 2 · 38 + 38 = 114 nT = 2 · 94 − 114 = 74 min(nA , nT ) = min(114, 74) = 74 Donde el p − value = 0,1355035 de la prueba es la probabilidad de observar el número de heterocigotos observados o una cantidad de heterocigotos más extrema, teniendo en cuenta las dos colas de la distribución. Manualmente podemos realizar el cálculo del p − valor, este se determina como la suma de todas aquellas probabilidades menores o iguales que la probabilidad de los heterocigotos observados, es decir, la probabilidad de los heterocigotos observados (38) 16 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA es 0.0533, pues la suma de todas las probabilidades menores o iguales que esta, donde quedarı́a la siguiente expresión. p−valor = 0,0002+0,0009+0,0032+0,0099+0,0253+0,0533+0,0275+0,0107+ 0,0034 + 0,0009 + 0,0002 u 0,1356 Por lo tanto no podemos rechazar la hipótesis nula de que haya equilibrio de HardyWeinberg. Observemos la distribución de los heterocigotos para ver su comportamiento, figura 2.5. Figura 2.5: Distribución de los heterocigotos Notamos que el soporte de la distribución son los números pares (0, 2, 4, . . . , min(nA , nB )) ya que el máximo de heterocigotos es par (74), si por el contario el máximo de heterocigotos fuese impar, entonces el soporte de la distribución serı́a (1, 3, 5, . . . , min(nA , nB )). 2.4.3. Test de Razón de Verosimilitud (LRT) El test de razón de verosimilitud es un test estadı́stico para tomar decisiones entre 2 hipótesis basadas en el valor de esta razón [6]. Esta razón no es más que el cociente de las verosimilitudes bajo cada una de las hipótesis planteadas, que sobre el tema que nos concierne, la verosimilitud de una muestra de conteos genotı́picos está dada por la distribución multinomial de la siguiente forma si estamos bajo la hipótesis alternativa: 2.5. MÍNIMA FRECUENCIA ALÉLICA (MAF) LA = n AA nAT nT T pnAA pAT pT T nAA , nAT , nT T 17 (2.10) Y el estimador máximo verosı́mil está dado por las frecuencias genotı́picas muestrales. Bajo la hipótesis de que existe equilibrio de Hardy-Weinberg, la verosimilitud tiene la siguiente expresión: L0 = n nAA , nAT , nT T nA 2nAA nA nT nAT nT 2nT T 2 2n 2n 2n 2n (2.11) Donde −2 veces el logaritmo de la razón de las verosimilitudes está dado por la expresión: 2 G = −2ln L0 LA = −4nAA · ln nA nA · nT nT − 2nAT · ln − 4nT T · ln 2np 4n2 pq 2nq (2.12) Y este estadı́stico tiene asintóticamente una distribución χ21 . Asintóticamente, el test de razón de verosimilitud es equivalente al test χ2 para HWE. Continuando con el ejemplo y sustituyendo los valores que anteriormente fueron calculados, obtenemos que: G2 = 2,195173 p − valor = 0,1384437 Para el ejemplo estudiado, vemos que los 3 test ( χ21 , Exacto y LRT) dan valores p muy parecidos, (0.1376, 0.1355, 0.1384) y que conducen a la misma conclusión. 2.5. Mı́nima frecuencia alélica (MAF) Dentro de una población, a los SNPs se les puede asignar una mı́nima frecuencia alélica, es decir, la menor frecuencia de alelos en un locus que se observa en una población en particular. Esto es simplemente la menor de las dos frecuencias de los alelos del SNP. Sean pA y pB las frecuencias alélicas de un SNP determinado. Por cada SNP conocemos que pA + pB = 1 por lo tanto se define como la mı́nima frecuencia alélica de la siguiente forma: 18 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA MAF = min (pA , pB ) ⇒ 0 ≤ MAF ≤ 0,5 (2.13) Para el ejemplo que hemos estado analizando tenemos que la menor frecuencia alélica la presenta pB = 0,39361 2.6. Coeficiente de endogamia (f ) La endogamia es la reproducción de la unión de 2 individuos relacionados genéticamente cuyo resultado es un incremento a favor de la homocigosis, que pueden aumentar las posibilidades de que la descendencia se vean afectados por los genes recesivos, en otras palabras, la endogamia puede dar lugar a un mayor número de expresión fenotı́pica de los genes recesivos dentro de una misma población. Este se computa como un porcentaje de posibilidades de que dos alelos sean idénticos por descendencia. Este porcentaje se denomina “coeficiente de endogamia”. El coeficiente de endogamia tiene la siguiente expresión [3]: f= Esp(fAT ) − Obs(fAT ) Obs(fAT ) =1− Esp(fAT ) Esp(fAT ) (2.14) es decir, es uno menos la frecuencia observada de los heterocigotos sobre lo esperado en equilibrio de Hardy-Weinberg. En la literatura, f también es conocido como el coeficiente de correlación intraclase y puede estimarse mediante el método de máxima verosimilitud de la siguiente forma: 4nAA nT T − n2AT fˆ = nA nB (2.15) Cuya varianza está dada por la siguiente expresión [2]: 1 f (1 − f )(2 − f ) V ar(fˆ) = (1 − f )2 (1 − 2 f ) + n 2 n pA (1 − pA ) Donde si f = 0 ⇒ V ar(fˆ) = (2.16) 1 n También conocemos que para 2 alelos, el coeficiente de endogamia tiene la siguiente relación con las frecuencias genotı́picas [2]: pAA = p2A + pA pT f pAT = 2 pA pT (1 − f ) pT T = p2T + pA pT f El dominio de estas frecuencias está dado por las siguientes expresiones: (2.17) 19 2.6. COEFICIENTE DE ENDOGAMIA (F ) 0 ≤ pAA ≤ pA 0 ≤ pAT ≤ min(2 · pA , 2 · pT ) (2.18) y para f −min(pA ,pT ) 1−min(pA ,pT ) ≤f ≤1 (2.19) Analizando estas expresiones en dependencia de los posibles valores que tome f podemos definir ciertos rangos: - Para f - Para f - Para f - Para f = 0 Equilibrio de Hardy-Weinberg = 1 Ausencia de Heterocigotos < 0 Exceso de Heterocigotos > 0 Déficit de Heterocigotos Estas condiciones determinan lo que justamente decı́amos en la introducción de este tema, en el caso de selección a favor de heterocigotos, es decir, donde el genotipo de mayor adaptabilidad es el heterocigoto y ambos homocigotos son afectados por la selección en contra, pero generalmente en proporciones muy diferentes, esta condición determina un equilibrio de las frecuencias alélicas muy especial. Por ejemplo en un caso extremo donde ambos homocigotos no pasen sus genes a la siguiente generación (letalidad completa a los homocigotos), es decir, sólo se reproducirı́an los heterocigotos entre sı́, generando una frecuencia alélica de 0.5 para los dos alelos. Las consecuencias de este hecho por ejemplo, serı́a que si un alelo es el responsable de una enfermedad genética, al pasar a la siguiente generación con una frecuencia de 0.5, pues el hecho de ser portador de esta enfermedad es del 75 %, es decir, la probabilidad de ser portador es del 75 % (50 % los heterocigotos, más 25 % homocigotos BB) entre los neonacidos. El test de chi-cuadrado para el equilibrio de proporciones de Hardy-Weinberg es equivalente a un test con H0 : f = 0. El estadı́stico X 2 se relaciona con el coeficiente de endogamia estimado mediante la expresión: X 2 = nfˆ2 Si proseguimos con el mismo ejemplo, tenemos: f =1− 38 = 0,1531539 44,87238 X 2 = 94 · 0,15315392 = 2,204875 (2.20) 20 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA Podemos comprobar que los valores obtenidos tanto en la prueba χ2 tradicional como usando el coeficiente de endogamia son casi idénticos. El coeficiente de endogamia es inestable cuando los valores esperados son próximos a cero y esto no es útil para alelos raros o muy comunes. 2.7. Potencia de las pruebas para detectar HWE Es posible calcular la potencia de las diferentes pruebas estadı́sticas para HWE. Con respecto a los datos estudiados en este proyecto, hay dos ámbitos en los que los cálculos de potencia son relevantes, y estos se describen brevemente a continuación. 2.7.1. Los cálculos de potencia de las pruebas clásicas para HWE El cálculo de potencia para la clásica prueba χ2 ha sido descrito por Weir [2, cap.3]. Bajo la hipótesis nula, el estadı́stico X 2 tiene una χ21 , bajo la hipótesis alternativa (Desequilibrio) el estadı́stico X 2 tiene una distribución no-central χ1,ν con un parámetro de nD2 no-centralidad ν = p2 (1−p 2 , donde n es el tamaño muetral, pA es la frecuencia alélica A) A y D es el parámetro de desequilibrio de Weir. La última está estrechamente relacionada con el coeficiente de endogamia presentado en la sección 2.6. Usando la distribución de χ2 no-central, para un tamaño muestral dado, nivel de significación y grado de desequilibrio D, la potencia del test χ2 puede ser calculada. Estos cálculos de potencia son, como se indica en Weir [2], aproximados y sólo son válido para pequeñas desviaciones de equilibrio. A la inversa, también se puede utilizar este resultado para calcular el tamaño de las muestras necesarias para obtener una potencia dada. Cálculos de potencia para la prueba exacta para HWE también son posibles, pero computacionalmente mucho más intensivas. Con el fin de calcular la potencia de la prueba exacta, la distribución del número de heterocigotos dado el menor conteo alélico, dado en la ecuación 2.9, bajo la hipótesis alternativa es necesaria. El grado de desequili2 /(P brio puede ser parametrizado por θ = PAB AA · PBB ). Bajo HWE, nosotros tenemos θ = 4. Valores de θ > 4 implica exceso de heterocigotos mientras θ < 4 significa déficit de heterocigotos. Por la selección de diferentes valores de θ, el grado de desequilibrio (el tamaño del efecto) puede ser especificado. Con esta parametrización, la distribución condicional de le número de heterocigotos, dado antes en la sección 2.4.2 puede ser reescrita en términos de θ. La potencia del Test Exacto dado los valores de θ y dado el menor conteo alélico, se puede calcular exactamente por la suma de las probabilidades de acuerdo con esta distribución condicional para todas las muestras que tienen un valor de p por debajo del nivel de significación especificado α. 21 2.7. POTENCIA DE LAS PRUEBAS PARA DETECTAR HWE 2.7.2. Los cálculos de potencia de una prueba de HWE para la asociación marcadores-enfermedad Lee [7] ha sugerido que una prueba para HWE se puede utilizar para la prueba de asociación genética entre el marcador y la enfermedad, utilizando una base de datos de los individuos afectados. Una población se suponı́a que debı́a empezar (al nacer) en HWE, pero después los genotipos AA, AB y BB pueden tener diferentes riesgos relativos φ1 (AB/BB) y φ2 (AA/BB). Estos factores de riesgo modifican las frecuencias de los genotipos y las frecuencias alélicas, lo que provoca desequilibrio de Hardy-Weinberg con parámetro de desequilibrio D = ((q(1 − q)/R)2 )(φ2 − φ21 ), donde q es la frecuencia alélica en la población inicial y R = q 2 φ2 + 2q(1 − q)φ1 + /(1 − q)2 , D será generalmente no-cero si φ2 6= φ21 . Lee [7] utiliza la raı́z cuadrada del estadı́stico X 2 clásico para HWE como un test estadı́stico y muestra que bajo equilibrio de Hardy-Weinberg, es decir, H0 : D = 0, es asintóticamente distribuido como una normal estandar. Estos resultados pueden ser utilizados para calcular el tamaño de la muestra y de la potencia para el test de HWE para la asociación de marcadores-enfermedad. Brevemente resumimos los tamaños de muestras calculados por Lee para obtener 80 % de potencia para frecuencias alélicas y los riesgos relativos dados de los diferentes modelos de enfermedad (aditivo, recesivo y dominante). Véase tabla 2.1 referida a la tabla 1 del artı́culo [7]. Estos resultados serán usados en la sección 5.9. γ 4.0 q Aditivo Recesivo Dominante 0.01 0.10 0.50 0.80 97,643 (0.80) 2,343 (0.80) 2,096 (0.80) 13,134 (0.80) 57,643 (0.80) 958 (0.79) 427 (0.80) 2,187 (0.80) 40,191 (0.80) 830 (0.80) 412 (0.81) 1,752 (0.80) 439,088 (0.80) 6,004 (0.80) 1,369 (0.80) 5,063 (0.80) 366,616 (0.80) 5,436 (0.80) 1,362 (0.81) 4,621 (0.80) 1,660,984 (0.80) 21,431 (0.80) 3,802 (0.80) 11,950 (0.80) 1,494,425 (0.80) 20,106 (0.80) 3,798 (0.80) 11,389 (0.80) 2.0 0.01 0.10 0.50 0.80 1.5 0.01 0.10 0.50 0.80 1,663,335 (0.80) 25,537 (0.80) 8,581 (0.80) 38,628 (0.80) Tabla 2.1: En esta tabla vemos que los tamaños de muestra son necesarios a fin de obtener una potencia del 80 % para la detección de asociación mediante la prueba de HWE 22 CAPÍTULO 2. CONCEPTOS BÁSICOS DE LA GENÉTICA 2.8. Importancia y aplicación del Equilibrio de HardyWeinberg El modelo de Hardy-Weinberg siendo una proposición teórica es muy valioso para evaluar los factores evolutivos que están operando en las poblaciones. Si una población no presenta estructura genética según este equilibrio, es porque están actuando algunos de los factores evolutivos. Según la relación entre homocigotos o heterocigotos, esperados y observados, se pueden deducir varias desviaciones. En términos de marcadores podemos remarcar 2 aplicaciones importantes en el Equilibrio de Hardy-Weinberg: I. Con él detectar errores de genotipado. II. Si un marcador está asociado a una enfermedad, se espera desequilibrio de HardyWeinberg (sección 2.7.2 y sección 5.9), es decir, el equilibrio de Hardy-Weinberg para un determinado marcador puede indicar que este marcador esté en un gen involucrado con la enfermedad. En estudios de Caso-Control, se espera desequilibrio para los Casos, pero no necesariamente para los Controles [7]. Capı́tulo 3 Descripción de la base de datos El cáncer colorrectal provoca 13.000 muertes anuales y es el primer tumor en incidencia y el segundo en mortalidad. Las causas exactas que lo producen se desconocen, aunque se han identificado factores de riesgo que favorecen su aparición, como los dietéticos (dieta rica en grasas y pobre en frutas y verduras), hábitos de vida no saludables, dolencias predisponentes, como pólipos o enfermedad inflamatoria intestinal (Crohn o colitis ulcerosa), historia previa de cáncer colorrectal, factores genéticos o historia familiar de cáncer de colon. 3.1. Motivación por esta Base de Datos El cáncer colorrectal, también llamado cáncer de colon, se produce debido al estilo de vida, aumento de la edad y sólo una minorı́a de casos asociados con trastornos subyacentes genéticos. Las personas con antecedentes familiares tienen mayor riesgo de tener la enfermedad y este grupo representa alrededor del 20 % de los casos con cáncer de colon. Una serie de sı́ndromes genéticos también están asociados con mayores tasas de cáncer colorrectal. El más Figura 3.1: Cáncer de Colon 23 24 CAPÍTULO 3. DESCRIPCIÓN DE LA BASE DE DATOS común de éstos es el sı́ndrome de Lynch que está presente en alrededor del 3 % de las personas con cáncer colorrectal. Otros sı́ndromes que están fuertemente asociados son: el sı́ndrome de Gardner, y la poliposis adenomatosa familiar (PAF) en el que el cáncer casi siempre se produce y es la causa de 1 % de los casos. Asimismo, desde el 2008 está disponible un catalogo de publicaciones de estudios sobre asociación de esta enfermedad en una gama del Genoma en la página web del Instituto Nacional de Investigación sobre le Genoma Humano. Hasta ahora hay reportado 30 SNPs coligados fuertemente al Cáncer de Colon, estos situados en diferentes genes por diferentes cromosomas, ası́ como aquellos alelos de mayor riegos [8]. Figura 3.2: Estadı́os del cáncer de Colon 3.2. Estructura de la base de datos La base de datos, objeto de estudio de este proyecto de fin de máster, fue proporcionada por el Doctor Victor Moreno del Hospital de Bellvitge. Se trata de una base de datos de 99 individuos, todos enfermos de cáncer de colon, donde fueron genotipados para 1000 marcadores genéticos (SNPs). Tenemos la siguiente estructura, la primera parte es la medida de la intensidad para la base nitrogenada A (IA), es decir, 1000 medidas de intensidades A para 99 individuos, la segunda parte es la medida de la intensidad para la base nitrogenada B (IB), con dimensión 1000 x 99 y la tercera parte, el genotipo, es el resultado de la clasificación y consta de 1000 SNPs para 99 individuos. Las partes de las medidas de las intensidades (IA e IB) están declaradas las variables como continuas y los SNPs son variables categóricas. Dentro de las variables categóricas, tenemos 912 SNPs politómicas y el resto son dicotómicas. Recordemos que existen 4 bases nitrogenadas y cuando nos referimos en este caso A y B es sólo de forma representativa ya que el algoritmo de clasificación automática asigna el genotipo de acuerdo con los alelos que tiene en cada gen y sus respectivos cromosomas. 3.2. ESTRUCTURA DE LA BASE DE DATOS La codificación de los SNPs en este caso ha sido establecida de la siguiente manera: 1 = AA para homocigotos 2 = AB para heterocigotos 3 = BB para homocigotos NA = Missing Data 25 Capı́tulo 4 Introducción a los Missing Data 4.1. Breve descripción del problema de los Missing Data en los SNPs Desde el enfoque estadı́stico los motivos de los Missing Data para una variable pueden ser muy diversos y pueden ir desde la total aleatoriedad hasta una fuerte dependencia de los valores reales de las variables [9], brevemente explicados en la introducción del trabajo. Para el punto de vista de las proporciones de Hardy-Weinberg esta pérdida de datos pueden traer consigo sesgos en las pruebas para HWE (χ2 o test exacto incorrectos) sobre todo si las tasas de pérdidas de datos es distinta para homocigotos y heterocigotos. 4.2. Missing Data en los SNPs Cuando la presencia de Missing Data ocurre por razones ajenas al investigador, es necesario establecer supuestos acerca de las causas que generaron estos Missing, contrastando la posibilidad de las hipótesis respecto al comportamiento de los datos observados. Un sistema por el cual los Missing Data se generan en los SNPs es mediante la mala clasificación de los genotipos a través de las medidas de la intensidad de cada base nitrogenada. El proceso de asignación del genotipo se realiza de la siguiente forma: o Se toma la intensidad de cada base nitrogenada. o Mediante un algoritmo de clasificación automática a través de combinación de Clustering y clasificación, el sistema asigna el genotipo en dependencia de las 27 28 CAPÍTULO 4. INTRODUCCIÓN A LOS MISSING DATA potencias de las intensidades de las bases nitrogenadas medidas y estas son proyectadas en un ’Call Plot’. o En general, una intensidad alta para A y baja para B corresponde a un homocigoto AA; una intensidad alta para B y baja para A, representa a un homocigoto BB y 2 intensidades entre media y alta para ambas intensidades, se califica como un heterocigoto AB. o Si el sistema detecta incoherencia entre las potencias de las intensidades clasifica el genotipo como perdido, es decir, declara un Missing Data. Figura 4.1. En el siguiente gráfico podemos observar un ejemplo de medida, clasificación y asignación de los genotipos para 99 individuos en un SNPs determinado. Figura 4.1: (a) Medida de Intensidad A, (b) Medida de Intensidad B, (c) Genotipado Los puntos marcados con una cruz son declarados según el sistema utilizado como Missing Data. Estas pérdidas de valores traen consigo imperfecciones negativas en el mapeo genómico y por ende problemas en el análisis comparativo del genoma. Se observa que hay una tendencia a que los missing se produzcan sobre todo en la frontera del grupo de heterocigotos. 29 4.3. TERMINOLOGÍAS 4.3. Terminologı́as Sea Ynxp una matriz de variables respuestas parcialmente observable en una muestra de tamaño n. En el presente caso, p = 1000 y n = 99, donde las columnas son: Y1 : SN P1 Y2 : SN P2 ··· : ··· Y1000 : SN P1000 Sea Znxq una matriz de covariables observables, para el problema de marcadores genéticos nosotros tenemos que q = 2000 donde las primeras 1000 columnas son referentes a la medida de intensidad A por cada SNP y el resto de las columnas a la medida de intensidad de B. IA1 : Intensidad de A para el SN P1 — IB1 : Intensidad de B para el SN P1 IA2 : Intensidad de A para el SN P2 — IB2 : Intensidad de B para el SN P2 ············ IA1000 : Intensidad de A para el SN P1000 — IB1000 : Intensidad de B para el SN P1000 4.3.1. Mecanismos de Respuestas en Marcadores Genéticos Esta terminologı́a está basada en el marco estándar dado por Rubin [10] y Little y Rubin [11]. Sean Yobs y Ymis que denotan las partes observadas y missing de Y , es decir, Y = (Yobs , Ymis ), nomenclatura conveniente pero imprecisa. En adición, asumimos cada unidad independiente i = (1, . . . , n) que representan los individuos y por cada j (SNP), j = (1, . . . , p), podemos definir el indicador de Missing Data Rnxp como una matriz binaria, descrita de la siguiente manera: ( 1 si Yij es observado Rij = 0 si Yij no se observa (4.1) Definiendo P (Rij = 0|Yij ) = P (Yij no observado|Yij ) = pij entonces Rij es sujeto a una distribución de probabilidad P (R|Y, ψ) regido por ψ. La parametrización de la distribución conjunta de R y Y puede expresarse mediante 3 modelos: p(Y, R|Z, θ, ψ) = p(Y |Z, θ)p(R|Y, ψ) =⇒ Modelos de selección p(Y, R|Z, θ, ψ) = p(Y |R, Z, θ)p(R|Y, ψ) =⇒ Modelos de patrones de mixtura p(Y, R|Z, θ, ψ) = p(Y |R, Z, θ, β)p(R|Y, ψ, β) =⇒ Modelos de parámetros compartidos 30 CAPÍTULO 4. INTRODUCCIÓN A LOS MISSING DATA Diferentes supuestos concernientes a la relación entre R con Yobs , Ymis y Z define diferentes tipos de mecanismos de respuestas. Explicaremos más adelante aquellos que son relevantes para nuestro estudio basado en los SNPs. Se distinguen tres tipos de mecanismos de pérdida de datos [12]: = MCAR (Missing Completely At Random): Si p(R|(Yobs , Ymis ), ψ) = p(R|ψ), es decir, el Missingness es independiente de la respuestas (Observado y Missing). = MAR (Missing At Random): Si p(R|(Yobs , Ymis ), ψ) = p(R|Yobs , ψ),es decir, el Missingness es independiente de la respuesta Missing dado los valores observados. = NMAR (Not Missing At Random): Si p(R|(Yobs , Ymis ), ψ) 6= p(R|Yobs , ψ), es decir, el Missingness depende de ambas respuestas, Observados y Missing. En términos de probabilidades un patrón de no respuesta se dice que es completamente aleatorio (MCAR) si las probabilidades de observación de algunas componentes y de no observación de otras no depende ni de los datos observados ni de los no observados. Si estas probabilidades solamente dependen de los datos observados entonces el patrón de no respuesta se dice que es aleatorio (MAR), sin embargo si estas probabilidades dependen de los valores no observados entonces el patrón de no respuesta se dice que es no ignorable (MNAR) y por lo tanto las inferencias no serán correctas si no se tiene en cuenta este hecho. Los dos conjuntos de parámetros θ y ψ se dicen ser distintos si: (1) desde una perspectiva frecuentista, el espacio paramétrico conjunto de (θ, ψ) es el producto cruzado cartesiano de los espacios paramétricos de θ y ψ. (2) desde una perspectiva Bayesiana, la distribución a priori conjunta de (θ, ψ) pueden ser factorizado en las distribuciones marginales a priori independientes para θ y ψ. Si θ y ψ son distintos, por [10] y [11] definimos que L(θ, ψ|(Yobs , Ymis ), R) p((Yobs , Ymis ), R|θ, ψ) donde p((Yobs , Ymis ), R|θ, ψ) puede ser reemplazada por p(Yobs |θ) ignorando el mecanismo de respuesta de missing y además si nuestras inferencias están basadas en θ entonces L(θ|Yobs ) p(Yobs |θ) Podemos especificar 5 mecanismos de respuestas aplicados a los marcadores genéticos, RSN Pj , de SNPs incompletos: (I) Mecanismo MCAR. Cuando por separados los valores observados y no observados por cada SNP tienen la misma distribución respecto a otras variables (otros marcadores o intensidades). 31 4.3. TERMINOLOGÍAS 1. Missing=Heterocigotos. La figura 4.1 insinua que la mayorı́a de los missing se produce en las fronteras del grupo de heterocigotos, esto sugiere que quizás el problema esté a la hora de asignar el heterocigoto debido a incordialidades entre las medidas de las intensidades. (II) Mecanismo MAR en Yobs . La probabilidad de asignación del genotipo en un SNP tiene cierta asociación con marcadores observados completamente desde el punto de vista multivariado o si analizamos el caso univariado que la probabilidad de este SNP dependa de los casos observados del mismo. (III) Mecanismo MAR en Z. La probabilidad de no respuesta está dada por las medidas de las intensidades de las bases nitrogenadas. 1. Mecanismo MAR diferenciado. Existe posibilidad de que el porcentaje de significativos no tiene porque ser exactamente igual con la intensidad de A y con la intensidad de B. Lo cual se podrı́a contrastar si estos porcentajes difieren de manera significativa. La prueba t-Student sugiere que existe esta diferencia. (Ver figura 5.7) (IV) Mecanismo MNAR. Que los SNPs con Missing estén condicionados por otros SNPs con Missing. Ambos modelos MAR pueden ser útiles combinados en un mecanismo MAR, p(R|Yobs , Z), es decir, condicionado a todos los datos observados. 4.3.2. Patrones de Missing Data De manera general podemos clasificar 2 tipos de patrones para las variables con missing data, donde 1 = Valores Observados y 0 = Valores No Observados. Patrones Monótonos (Dropouts): Una secuencia de valores observados y a partir de una determinada posición o tiempo, esta secuencia se deja de observar hasta el final. Como se muestra en la figura 4.2 Figura 4.3: Un patrón no monótono de Missing Data Figura 4.2: Un patrón monótono de Missing Data Patrones no Monótonos: Una secuencia de observaciones donde existen missing en diferentes posiciones o tiempos, sin seguir una pauta, es decir, entre observaciones podemos tener valores no observados. Figura 4.3. 32 CAPÍTULO 4. INTRODUCCIÓN A LOS MISSING DATA Problemas que surgen con los patrones no monótonos Los patrones no monótonos complican mucho la modelación, estimación y el proceso para la imputación de los Missing Data [10]. La modelación es mucho más difı́cil debido a que los diagnósticos estándares no son apropiados, el mismo problema surge con la estimación ası́ como a la imputación de los valores perdidos incluso con valores conocidos del parámetro debido a la necesidad de encontrar la distribución condicional no explı́citamente formulada en la modelación. Estos problemas están basados en modelos explı́citos aunque análogamente surgen estos problemas cuando usamos modelos implı́citos para la imputación de patrones no monótonos, por lo tanto es necesario desarrollar herramientas buenas para el caso de estos patrones. Existen 5 soluciones generales [10]: I. Descartar algunos datos para crear un patrón monótono. II. Asumir independencia condicional entre bloques de variables para crear patrones monótonos. III. Usar un modelo explı́cito, analı́ticamente tratable pero posiblemente no totalmente adecuado. IV. Aplicar iterativamente métodos para patrones monótonos con modelos explı́citos. V. Usar el algoritmo de SIR (Sampling/Importancia Resampling) para modelos explı́citos apropiados. 4.4. Teorı́a general de la imputación El objetivo de cualquier técnica de imputación es producir un conjunto de datos completos, para ser tratados usando métodos inferenciales de datos completos [13]. Dos tipos de métodos de imputaciones son los más usados: Imputación Simple e Imputación Múltiple. En [10] y [11] los métodos de imputación se clasifican como se muestra a continuación: ♥ Análisis de datos completos (listwise) ♥ Análisis de datos disponibles (pairwise) ♥ Imputación por medias no condicionadas ♥ Imputación por medias condicionadas mediante métodos de regresión ♥ Máxima Verosimilitud (MV) 33 4.4. TEORÍA GENERAL DE LA IMPUTACIÓN ♥ Imputación Múltiple (MI) Las bondades de los procedimientos de imputación no deben valorarse por el sólo hecho de que permiten completar información para hacer inferencia sobre hipótesis y análisis de regresión. Los criterios para evaluar la eficacia de un método estadı́stico fueron establecidos por Neyman y Pearson (1933) y Neyman (1937) y guardan relación con el error cuadrático medio (ECM) y no sólo con el sesgo del estimador [14], es decir, si dado un SNP que contenga Missing Data y si la imputación que se realiza es la adecuada, entonces supongamos que analizamos el coeficiente de endogamia f de este SNP, por tanto el estimador f̂ será cercano al verdadero valor del parámetro f en muestras repetidas. De esta manera se logra minimizar el sesgo, la varianza y la desviación estándar de f̂, de otra manera, el sesgo y la varianza se combinan en la medida ECM que se computa como el promedio de la distancia entre (f̂ − f)2 sobre muestras repetidas; por tanto el ECM (f ) = Sesgo(fˆ)2 + V ar(f̂) [15]. Por lo que podemos decir que, el sesgo, la varianza y el ECM describen el comportamiento de un estimador. El error estándar (se) deberı́a ser parecido a la desviación estándar, en tanto que los intervalos de confianza deben incluir al verdadero valor del parámetro f con probabilidad cercana a la tasa nominal, por lo que obtendrán intervalos más pequeños lo cual reduce la probabilidad de error tipo II [14]. 4.4.1. Imputación Múltiple (IM) Imputación Múltiple es una técnica que reemplaza cada Missing con 2 o más valores aceptables representando una distribución de probabilidades. Véase figura 4.4. La idea fue originalmente propuesta por Rubin en 1977. La estrategia a seguir mediante esta técnica se describe a través de 4 pasos: Figura 4.4: Imputación Múltiple 1. Especificar la densidad posterior predictiva p(Ymis |X, R), donde X es un conjunto de variables predictoras, dado el mecanismo de no-respuesta, p(R|Y, Z) y el modelo de datos completos p(Y, Z). 2. Elaborar las imputaciones a partir de esta densidad para producir m conjuntos de datos completos. 3. Desarrollar m análisis de datos completos en cada matriz de datos ccompletados. 4. Realizar la combinación de los m análisis (el “Pooling”) del paso anterior resultando finalmente los estimadores para los parámetros y sus varianzas. 34 CAPÍTULO 4. INTRODUCCIÓN A LOS MISSING DATA Describiendo los pasos anteriores según el marco dado por [10] y [11]. Sea θ̂l los estimadores de datos completos de un parámetro de interés, por ejemplo el coeficiente de endogamia en genética, Ŵl , l = 1, . . . , M , sus respectivas varianzas asociadas para θ calculadas desde las M imputaciones repetidas bajo un modelo determinado. El análisis del conjunto de datos obtenidos a través de imputación múltiple es bastante directo. La combinación estimada para θ es: θ̄M = M X θ̂l M l=1 La variabilidad asociada con f tiene 2 componentes: el promedio de la varianza intraimputación: M X Ŵl W̄M = (4.2) M l=1 y la componente entre-imputación: PM − θ̄M )2 M −1 l=1 (θ̂l BM = (4.3) La varianza total asociada con θ̄M es TM = W̄M + M +1 · BM M donde (MM+1) es un ajuste para un M finito. Con el scalar θ, la distribución de referencia es una t-student −1/2 (θ − θ̄M )TM :tv (4.4) donde los grados de libertad v = (M − 1) 1 + M W̄M · M + 1 BM 2 (1−γ) M el resultado W̄ donde γ es la fracción de información missing BM estima la cantidad γ sobre θ debido a la no respuesta. Esta fracción está dada por la expresión: γM 2 rM + v+3 = rM + 1 donde rM es el incremento relativo de la varianza B cuya expresión es rM = M + 1 BM M W̄M Para la realización de este estudio nuestro parámetro de interés será el coeficiente de endogamia (f), donde en la sección 2.6 se describió una breve reseña sobre su teorı́a, 35 4.4. TEORÍA GENERAL DE LA IMPUTACIÓN importancia y aplicación. Otra cuestión importante que debemos destacar es cuántas veces debemos imputar, ya que este hecho es muy fundamental para la eficiencia de nuestros estimadores. Rubin [10] señala que para tasas de respuestas inusualmente altas sólo requiere generar entre 5 y 10 imputaciones, aunque afirma que el método de Imputación Múltiple es capaz de generar resultados robustos con un número más pequeño de iteraciones. En nuestro trabajo usaremos m = 10 para una mejor convergencia y eficiencia. Esta eficiencia γ −1 puede ser calculada como (1 + M ) , donde γ es la fracción estimada de la información Missing, donde tanto en [10, p.114] y [16, p.110] podemos encontrar la tabla que a continuación exponemos: γ m 2 3 5 10 20 0.1 95 97 98 99 100 0.3 87 91 94 97 99 0.5 80 86 91 95 98 0.7 74 81 88 93 97 0.9 69 77 85 92 96 Tabla 4.1: Eficiencia relativa ( %) de la estimación mediante Imputación Múltiple por número de imputaciones y fracción de información Missing 4.4.2. Modelos de Imputación Varios modelos de imputación han sido desarrollados en diferentes contextos. En general la estrategia para construir modelos de imputación caen en 2 categorı́as [17], [18]. 1. Modelación Conjunta: El enfoque de la Modelación Conjunta implica especificar una distribución multivariada para los Missing Data y elaborar las imputaciones desde sus distribuciones condicionales mediante técnicas de simulación de Monte Carlo vı́a Cadenas de Markov. Dentro de la modelación conjunta encontramos los Modelos de Localización General, que serán descrito más adelante. Estos métodos comienzan por especificar la densidad multivariada paramétrica para los datos, dados los parámetros del modelo. Bajo una apropiada distribución a priori de los parámetros, es posible derivar el submodelo adecuado por cada patrón de Missing Data, para el cual las imputaciones son creadas. El enfoque de modelación conjunta en teorı́a es buena pero puede carecer de la flexibilidad necesaria para representar estructuras de datos complejas que surgen en muchos estudios, en tal caso, la estrategia de modelación conjunta es difı́cil de implementar debido a que las especificaciones de las distribuciones multivariadas no son suficientemente flexibles para acomodar estas funciones. 36 CAPÍTULO 4. INTRODUCCIÓN A LOS MISSING DATA 2. Imputación Múltiple de Regresión Secuencial, SRMI: también referido como Imputación Múltiple a través de Chained Equations [19]. Los datos multivariados son caracterizados por modelos condicionados separados por cada variable incompleta. Esto es, el modelo de imputación es especificado separadamente por cada variable, con otras variables como predictoras. En cada paso de este algoritmo, las imputaciones son generadas por los valores Missing de una variable, estos valores imputados son usados en la imputación de la próxima variable y este proceso se repite hasta que se alcanza la convergencia. Comparando el algoritmo SRMI con el enfoque de la Modelación Conjunta, una caracterı́stica atractiva de SRMI es que es relativamente fácil de acomodar las caracterı́sticas de datos complejos en los modelos de regresión univariante. Las variables dicotómicas se pueden modelar mediante regresión logı́stica y las variables categóricas con más de dos categorı́as a través de modelos politómicos. La construcción de estos modelos de regresión pueden seguir las pautas comunes de un modelo de regresión aplicado a los datos disponibles. 4.4.3. Modelo de Localización General (GLM) En la práctica generalmente los datos envuelven variables de diversos tipos. Existen métodos multivariados que relacionan estos datos mixtos. El Modelo de Localización General discutido por Little y Schluchter (1985) es uno de ellos, cuyo desarrollo se basa en la relación entre las funciones de verosimilitudes de estas variables. En presencia de Missing Data este método proveé un relativo y computacionalmente simple método Expectation-Maximization (EM) ası́ como otros métodos, por ejemplo Data Augmentation (DA). El marco de la terminologı́a está basada en Schafer [15] y Rubin y Little [11]. Describiendo y definiendo la figura 4.5 sea W1 , W2 , . . . , Wp un conjunto de variables categóricas y Z1 , Z2 , . . . , Zq un conjunto de variables continuas. Si estas variables son recolectadas por una muestra de tamaño n, el resultado es una matriz de nx(p+q) cuya nomenclatura podemos definirla de la siguiente forma: Y = (W, Z), donde W representa la parte categórica y Z la parte continua. Figura 4.5: Conjunto Datos con Missing Data 4.4. TEORÍA GENERAL DE LA IMPUTACIÓN 37 Verosimilitud de los datos completos Podemos escribir la función de verosimilitud de los datos completos como el producto de las verosimilitudes de la siguiente manera: L(θ|Y ) L(π|W ) · L(µ, Σ|W, Z) (4.5) Esta fórmula también puede ser factorizada desde el enfoque de la inferencia Bayesiana el cual simplifica la estimación de los parámetros, asumiendo independientemente distribuciones a priori para π y (µ, Σ) cuyos conjuntos también serán independientes en sus distribuciones a posteriori. Schafer en su libro [15] explica los 2 algoritmos que aplicaremos para el estudio de los Modelos de Localización General, los métodos EM y DA. El método EM es muy bien conocido, cuya idea general se basa grosso modo en repetir estos 2 pasos: 1. Expectación o E-Step, que no es más que encontrar la LogVerosimilitud esperada de θ. 2. Maximización o M-Step, en el cual θ(t+1) es encontrado maximizando la LogVerosimilitud esperada de θ. El proceso termina cuando |θ(t+1) − θ(t) | < T OL donde la condición de parada T OL o llamado también criterio de prueba para convergencia está dada por el investigador. En genética usualmente encontramos 10−6 o 10−7 . En términos iterativos podemos decir que esta metodologı́a consta de 5 pasos a seguir: 1. Reemplazar los valores missing por los valores estimados. 2. Estimar con la nueva muestra los parámetros. 3. Reestimar los valores missing asumiendo que el nuevo parámetro estimado es correcto 4. Reestimar los parámetros dado el paso anterior. 5. Repetir los pasos 3-4 hasta el criterio de prueba de convergencia. Por otro lado, la idea general del método Data Augmentation surge naturalmente en problemas de Missing Data [20], [21], cuyo fundamento está basado en un esquema de aumentar los datos observados, como bien lo indica su nombre. Este método en conjunto con el método EM tiene grandes ventajas para la solución de los problemas de máxima verosimilitud. En situaciones cuando la verosimilitud no puede ser aproximadamente cercana a una verosimilitud normal, los estimadores máximos verosı́miles y los errores estandar asociados no suelen dar inferencias válidas. 38 4.5. CAPÍTULO 4. INTRODUCCIÓN A LOS MISSING DATA Análisis de sensibilidad Un malentendido común acerca de la imputación múltiple es que está restringida a MAR [19, p.52]. Si bien es cierto que las técnicas de imputación comúnmente asumen MAR, la teorı́a de la imputación múltiple es completamente general y se aplica también a MNAR. Una alternativa sensible es la creación de una serie de escenarios posibles e investigar las consecuencias de cada una de ellas sobre las inferencias finales. En Rubin [10] existen un número de técnicas básicas. En nuestro estudio realizaremos aquellos modelos que expusimos en el punto IV de la sección 4.3.1 como modelos contraparte de los modelos MAR aplicados, es decir, tomaremos aquellos modelos MAR y les incluiremos otros SNPs que no se observaron completamente. En adición sobre los modelos MAR, llevamos a cabo otro enfoque al análisis de sensibilidad utilizando estrategias alternativas de modelado. 4.6. Metodologı́a de nuestro estudio de los Missing Data en el contexto de HWE Para el estudio de los Missing Data es necesario confeccionar un esquema para el diagnóstico de los modelos de imputación ası́ como el chequeo de estos. En nuestro estudio usamos diferentes paquetes de modelación, sumarizando en cada uno varios pasos envueltos en el proyecto de la imputación múltiple. Este proyecto tiene implı́cito diferentes pasos claves: (a) Análisis del Equilibrio de Hardy-Weinberg de las variables observadas completamente. (b) Para las variables incompletas hicimos un estudio sobre los patrones de Missing Data ası́ como la comprobación del mecanismo de Missing seguido por estas. (c) Hicimos apropiados supuestos para el mecanismo de Missing Data, explicados en la enumeración de la sección 4.3.1 referente a los mecanismos de respuestas aplicados a los marcadores genéticos. (d) Identificamos las variables a ser incluidas en el proceso de imputación. La estrategia general será explicada en la sección 5.4.2 mediante 2 criterios. (e) Construimos los modelos de imputación basados en modelos conocidos, factibles y sofisticados, implementados en el software R y explicados en la sección 4.4.2. (f) Seguidamente se realizó un diagnóstico de las imputaciones, ası́ como el análisis del sesgo producidos por los casos completos respecto a las imputaciones. 4.6. METODOLOGÍA DE NUESTRO ESTUDIO DE LOS MISSING DATA EN EL CONTEXTO DE HWE 39 (g) Post-Imputación se calculó cada componente del Pooling y se realizó el análisis de sensibilidad por cada coeficiente de endogamia estimado para los SNPs escogidos. (h) Como objetivo final se realizó el estudio para el Equilibrio de Hardy-Weinberg. Capı́tulo 5 Análisis de los resultados 5.1. Estadı́stica Descriptiva de los SNPs completos Comenzaremos todo el análisis de lo que hemos expuesto en los capı́tulos 2 y 4. Iniciaremos la estadı́stica descriptiva para el análisis de los SNPs sin Missing Data. El principio de Hardy-Weinberg se puede aplicar de dos maneras, ya sea una población que supone que tiene proporciones de Hardy-Weinberg, en la que las frecuencias de los genotipos pueden calcularse, o si las frecuencias de los tres genotipos son conocidos, donde pueden ser probados que las desviaciones son estadı́sticamente significativas. En nuestro caso lo reduciremos al segundo objetivo. Existen 376 SNPs de datos completos a los cuales les aplicaremos las pruebas estadı́sticas descritas en el capı́tulo 2. Comenzamos con la representación gráfica de la Mı́nima Frecuencia Alélica, gráfico 5.1, que se refiere a la frecuencia con la que el alelo menos común de los SNPs se produce en una población dada, más general, debido a las variaciones entre las poblaciones humanas, un alelo de mı́nima frecuencia en un SNP que es común en un grupo geográfico o étnico puede ser mucho más raro que en otros grupos. Figura 5.1: MAF El hecho de que haya un pico cerca del 0 indica que hay relativamente más marcadores con frecuencias alélicas extremas, que es lo más común en este fenomeno, ya que 41 42 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS se suelen observar en bases de datos de SNPs, como una ley de validez empı́rica en un lugar del genoma. Realicemos los 3 Tests: el test de χ2 , el test Exacto y el test LRT para comprobar si existe el equilibrio de Hardy-Weinberg o no entre las proporciones. Debido a la dimensionalidad de los datos, no expondremos los valores de los test, sólo realizaremos los Q − Q Plots y el número de SNPs significativos por cada test. Existen varias representaciones gráficas que permiten explorar el grado de cumplimiento del HWE de los marcadores; el qqplot, el diagrama ternario y diagramas bivariantes de frecuencias genotı́picas. Chi−square Q−Q Plot HWE_LRatio_test 80 100 Chi−square Q−Q Plot HWE_Chisquare_test ● 60 ● ● ● ● ● 40 ● ● ● ● 20 ● ● ● ● ● ● ● 40 Sample Chisquare quantiles 60 ● ● 20 20 40 60 80 100 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 0 20 Theoretical Chisquare quantiles (a) Q-Q plot Chi-Square 15 ● ● ● ● ● ● ● 10 −log(Observed p value) 60 (b) Q-Q plot Log-LikeliHood Ratio Q−Q plot HWE_Exact_test ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 0 40 Theoretical Chisquare quantiles 5 0 0 ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 0 0 Sample Chisquare quantiles 80 ● 5 10 15 −log(Expected p value) (c) Q-Q plot H-W Exact Test Figura 5.2: Q-Q Plots de los 3 test 80 5.1. ESTADÍSTICA DESCRIPTIVA DE LOS SNPS COMPLETOS 43 Referente a los Q−Q plots de los valores p de cualquiera de las pruebas de equilibrio de Hardy-Weinberg (gráfico 5.2c), son los valores p empı́ricos versus los cuantiles de los p valores teóricos de la distribución que siguen cada una de las pruebas. Para hacer este Q − Q plot más comparable con los del test χ2 y LRT, es mejor usar −10log10 de los valores p. Ası́ mismo los Q − Q plots para las pruebas χ2 y LRT (gráficos 5.2a-5.2b), muestran los cuantiles muestrales versus a los cuantiles teóricos, permitiendo observar cuán cerca está la distribución de un conjunto de datos a la distribución de referencia bajo la hipótesis nula. Podemos observar que según los 3 test que exponemos, existe homogenidad en cuanto a la cantidad de SNPs significativos, es decir, se encontró por cada test evidencia de que podamos rechazar la hipótesis de que estén en equilibrio de Hardy-Weinberg. El número de SNPs significativos resultó ser entre 15-23 SNPs en fuerte desequilibrio para las 3 pruebas. Los Q − Q plots de los marcadores completamente observados parecen a primera vista indicar que el equilibrio de Hardy-Weinberg no se cumple para esta base de datos ya que hay 23 SNPs muy significativos. Sin embargo, el número esperado de resultados significativos es, al nivel del 5 %, aproximadamente 0,05 · 376 ≈ 19 SNPs y es del mismo orden en magnitud que el número de significativos observados. Si empleamos un nivel de significación del 1 %, se esperan unos 4 SNPs significativos, mientras que encontramos 23 SNPs. Eso pone de manifiesto que los marcadores que salen significativos tienden a ser muy siginificativos, sea por error de genotipado o por otra causa. Como habı́amos comentado, existen otras 2 vı́as de analizar el equilibrio entre las proporciones de Hardy-Weinberg que son mediante un plot ternario y diagramas de Dispersión, representados en la figura 5.3. Las composiciones genotı́picas de 3 vı́as (AA, AB,BB), se pueden exponer en un diagrama ternario y además se puede representar la región de aceptación de las diferentes pruebas de equilibrio de Hardy-Weinberg en el mismo [22]. Esto permite una prueba gráfica de un gran conjunto de marcadores (SNPs por ejemplo) para HWE, el significado (o no) de la prueba para HWE se puede deducir de la posición del marcador en el plot ternario. Diferentes pruebas estadı́sticas para HWE se puede hacer gráficamente: la prueba ordinaria de Chi cuadrado, la prueba de Chi cuadrado con corrección de continuidad y la prueba exacta de Levene-Haldane. En el Plot Ternario de la figura 5.3a la región de confianza está basada en la prueba Chi cuadrado ordinaria. Los scatterplots realizan diagramas de dispersión de la frecuencia de AB o BB en 44 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS comparación con la frecuencia de AA y representan una curva que indica la condición de equilibrio de Hardy-Weinberg. (a) Plot Ternario (b) Scatterplot (c) Scatterplot Figura 5.3: Diagrama ternari y ScatterPlots de las frecuencias genotı́picas En todos los gráficos estudiados se observa que los marcadores tienden acercarse a la curva de HWE, en el diagrama ternario, figura 5.3a, se encontraron 23 SNPs fuera de la región de aceptación, lo cual está en correspondencia con los marcadores expuestos en la figura 5.2a. Como información adicional, podemos decir que aquellos marcadores significativos, que se encuentran por debajo de la curva presentan un déficit de heterocigotos y por encima de la curva un exceso de heterocigotos. Para los datos estudiados, los marcadores en desequilibrio suelen presentar una falta de heterocigotos. 45 5.2. INSPECCIONANDO LOS MISSING DATA 5.2. Inspeccionando los Missing Data Comenzaremos el estudio del comportamiento de los missing en la base de datos Cáncer de Colon. Para esto veremos la tabla de patrones de Missing. Examinaremos los patrones de Missing a través de ambas intensidades A y B, donde el número de Missing puede ser contado y visualizado de las siguientes maneras. Realizaremos inicialmente un plot de frecuencias de los Missing por cada SNP en la figura 5.4. Observamos que existen: # mis Frec: SNPs 0 376 1 199 2 105 3 59 4 44 5 29 6 27 7 25 8 24 9 10 El resto de los SNPs presentan más de un 10 %, lo cual analizaremos para averiguar en qué tipo de mecanismos de pérdida de datos estamos presentes. En total hay 3873 missings en toda la Base de Datos representando estos el 4 % de toda la información genotı́pica. Figura 5.4: Conteo de Missing por SNPs El diagrama de frecuencias de los Missing por Individuos lo podemos observar en la figura 5.5. Observamos que existen: # mis Frec: Ind 19 4 27 3 32 2 19 2 El resto de los individuos varı́an en números de missings donde todos presentaron missings, es decir, si eliminamos los individuos con casos missing perdemos la muestra completa. Figura 5.5: Conteo de Missing por Individuos 46 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Hemos indagado anteriormente sobre el comportamiento de los missing de nuestros datos, pero no hemos explicado el por qué se generan para esta base de datos en concreto. Como describimos en el capı́tulo 4.2, una de las causas de los Missing Data en genética es la mala clasificación del genotipado a la hora de la asignación del genotipo mediante las medidas de las intensidades, comenzaremos exponiendo 4 ejemplos para ver la relación entre las intensidades y la asignación del genotipo. (a) SNP 52 (b) SNP 71 (c) SNP 80 (d) SNP 125 Figura 5.6: Diagramas bivariantes de intensidades para 4 SNPs La figura 5.6 muestra 4 ejemplos de “Call Plot ” para 4 SNPs de la base de datos. En el gráfico 5.6a para el SNP 52, observamos el caso ideal de clasificación de genotipado pues no hay presencia de casos perdidos y hay una buena separación de los 3 genotipos, 47 5.2. INSPECCIONANDO LOS MISSING DATA para el SNP 71, gráfico 5.6b, se observa que para bajas intensidades de B, el sistema clasificó a todos los individuos como AA y los SNPs 80, gráfico 5.6c y 125, gráfico 5.6d, representan casos con missings, donde los individuos entre la nube de heterocigotos y homocigotos son clasificados como Missing. Análisis de los patrones de Missing Data Podemos observar los patrones tanto por SNPs como por Individuos. Por individuos sólo se pudo reducir a 92 patrones de 99 (No de individuos) y por SNPs 451 patrones de 1000 (No de SNPs). En ambos los patrones son no monótonos. Vemos que existen patrones repetidos, por ejemplo, si analizamos desde el espacio de los individuos, sólo 11 individuos compartieron iguales patrones, 4 de un tipo, 3 de otro, 2 y 2, coincidiendo entre ellos tanto en números de missing como en sus posiciones respectos a los SNPs, el resto cada uno tenı́a su propio patrón particular. En mismo análisis podemos ver en el espacio de las variables SNPs, hubo 376 patrones completos, 16 SNPs con un sólo missing en la posición del individuo 3, etc. El individuo que más aportó missing, fue el individuo 87, con un total de 117 missing de 1000 SNPs analizados y el que menos el individuo 39 con sólo 8 missing. Para el caso de los SNPs, el que más aportó fue un SNP con un total de 94 missing de 99 individuos, véase figura 5.4. En el estudio hemos eliminados aquellos SNPs con más de 50 % de la información perdida, siendo un total de 5 SNPs. 5.2.1. Mecanismo de Patrones de Missing Data Para el estudio del comportamiento de los Missing y para la comprobación sobre cuál mecanismo de Patrones de Missing Data tenemos presente, usamos 2 pruebas, las pruebas t de Student y la T 2 de Hotelling. Estos métodos son simples procedimientos para comparar en una misma variable las medias de la distribución de los casos observados y los casos Missing en el caso del t de Student ası́ como para la prueba T 2 -Hotelling pero vista como vector de p componentes, donde tales tests son útiles para comprobar si son MCAR las variables pero tienen ciertas limitaciones en cuanto a la potencias si la muestra de los casos incompletos es pequeña [11]. Por lo tanto, escogimos los SNPs que contuvieran entre un 10 % y un 50 % de Missing, alcanzando 97 SNPs. Se calculó el intervalo de confianza del 95 % para la diferencia de medias µobs − µnoobs , tanto para la intensidad A (figura 5.7a) como para la intensidad B (figura 5.7b) Tests Univariado y Multivariado aplicado a 97 SNPs Intensidad A T.T est T 2 Hotelling Intensidad B 61 Significativos 50 Significativos 74 Significativos 48 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS ● ● ● ● ● ● ● ● ● ● ●● ● ● −1500 ● 20 40 60 Marcadores (a) CI: Intensidad A ● ● 80 ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ●● ● ●● ● ● ● ●● ●● ● ●● ● ● ●● ● ● ● ● ●● ●● ●● ● ● ● ● ● ●● ●● ●● ●● ● ● ●● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ●● ● ● ● ● ● ● ●●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ●● ● ● ● ● ● ● ● ● 0 1500 ● ● ● ● ● 500 ● −1500 −500 0 CI 500 1000 ● ● ● ● 0 1500 ● ● ● 1000 ● ● ● −500 ● T.Test−−Intensidad B CI 2000 T.Test−−Intensidad A 100 ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ●● ● ● ● ●● ● ● ●●● ● ● ● ●●● ● ● ● ● ● ● ● ● ● ● ● ● ●●● ● ● ● ● ● ● ●● ● ● ●● ● ● ● ● ● ●● ●● ● ●● ● ●●● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ●● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ●● ● ● ● ● ● ● ● ● ● ●● ● ● ● ●● ● ● ● ●● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● 0 20 40 60 80 100 Marcadores (b) CI: Intensidad B Figura 5.7: Pruebas Mecanismo de los Patrones Missing Data Para muchos SNPs la diferencia de medias resulta ser significativas. Para los 61 SNPs de los 97 (aproximadamente 63 %) se descarta igualdad de intensidad de A para genotipos observados y no observados. Para intensidad B, se encontraron 50 SNPs significativos (aproximadamente 52 %). Si los datos fueran MCAR, hubiéramos esperado solamente un 5 % de signficativos por efecto del azar. Los porcentajes de significativos son mucho más grandes, indicando esto que podemos rechazar la hipótesis de que nuestros datos de forma global no son MCAR, por lo tanto asumimos que estamos bajo el modelo MAR. Las pruebas para igualdad de vectores de medias con la T 2 Hotelling tienen un porcentaje de significativos todavı́a más elevado y también ponen de manifiesto que los datos multivariados no son MCAR . La figura 5.7 muestra los intervalos de confianza del 95 % para la diferencia teórica entre las medias de los intervalos de los individuos para los genotipos observados y no observados. Como se puede observar en el gráfico para un porcentaje sustancial de los marcadores (>> 5 %) el intervalo no cubre el cero, indicando gráficamente que los datos no son globalmente MCAR. Otra forma de ver el estudio de los patrones es calculando el número de observaciones por patrones de missing para todos los pares de variables, ası́ como el número de observaciones perdidas, es decir, ver los patrones desde el punto de vista por ejemplo: (SN P 784obs , SN P 235obs ) y (SN P 784miss , SN P 235miss ), también se pudieran analizar 2 patrones más, que serı́an: (SN P 784obs , SN P 235miss ) y (SN P 784miss , SN P 235obs ), en general existen 4 tipos de patrones por pares de variables, aunque sólo expondremos los 2 primeros. 49 5.2. INSPECCIONANDO LOS MISSING DATA Comencemos el estudio realizando los cálculos descritos en el párrafo anterior, tomando una representación de 10 SNPs de los 97, es decir, cogamos aleatoriamente 10 SNPs que tengan entre un 10 % y 50 % de observaciones Missing. SNP645 SNP294 SNP9 SNP194 SNP297 SNP417 SNP680 SNP594 SNP510 SNP197 SNP645 70 53 51 52 52 53 56 55 55 54 SNP294 53 70 51 50 54 55 58 58 53 54 SNP9 51 51 70 51 54 52 56 54 55 57 SNP194 52 50 51 73 54 59 58 58 58 56 SNP297 52 54 54 54 75 58 58 58 59 61 SNP417 53 55 52 59 58 77 59 60 63 59 SNP680 56 58 56 58 58 59 78 63 64 58 SNP594 55 58 54 58 58 60 63 78 59 60 SNP510 55 53 55 58 59 63 64 59 78 60 SNP197 54 54 57 56 61 59 58 60 60 78 Tabla 5.1: SNPs que menos Observaciones aportaron La tabla 5.1 se interpretarı́a como aquellos SNPs que menos observaciones tuvieron. En la diagonal se observan la cantidad de observaciones en cada SNP y en los demás elementos de la matriz observamos aquellas coincidencias de todos los valores observados entre los SNPs por individuos. Analicemos por ejemplo el SNP 645 que menos observaciones presentó y crucémoslo con el SNP 197. En la intercepción de ambos SNPs se muestran 54 observación coincidente, esto quiere decir, que entre los 2 SNPs, tuvieron por filas, 54 observaciones coincidentes, expresándolo de otra manera, hubo 54 individuos, al que se le observaron los SNPs 645 y 197 conjuntamente y se pudo obtener el genotipo en dichos locus, es decir, hubo respuesta y ningún Missing para ambos SNPs. SNP645 SNP294 SNP9 SNP194 SNP297 SNP417 SNP680 SNP594 SNP510 SNP197 SNP645 29 12 10 8 6 5 7 6 6 5 SNP294 12 29 10 6 8 7 9 9 4 5 SNP9 10 10 29 7 8 4 7 5 6 8 SNP194 8 6 7 26 5 8 6 6 6 4 SNP297 6 8 8 5 24 5 4 4 5 7 SNP417 5 7 4 8 5 22 3 4 7 3 SNP680 7 9 7 6 4 3 21 6 7 1 SNP594 6 9 5 6 4 4 6 21 2 3 SNP510 6 4 6 6 5 7 7 2 21 3 SNP197 5 5 8 4 7 3 1 3 3 21 Tabla 5.2: SNPs que más Missing aportaron La tabla 5.2 por el contrario a la tabla 5.1 esta refleja aquellos SNPs que más Missings aportaron a nuestro ejemplo. Como mismo explicamos, en la diagonal vemos la cantidad de valores perdidos por cada SNP y por encima y debajo de esta, aquellos Missings coincidentes entre los individuos. Sigamos con el ejemplo anterior. 50 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Figura 5.8: Proporción de Missing y Combinaciones El SNP 645 presenta 29 observaciones perdidas de 99 individuos registrados, el SNP 197 tiene 21, pero entre ellos hay 5 individuos que coinciden en cuanto a que no se pudo obtener el genotipo en esa posición de esos SNPs. De manera general, esta tabla representa los datos Missing por SNPs y la relación entre aquellos individuos según la cantidad de Missings coincidentes por SNPs. La figura 5.8 resume lo descrito en las tablas 5.2 y 5.1. En el panel izquierdo vemos el porcentaje de Missings por cada SNP y que está en correspondencia con lo descrito en la tabla 5.2 y en el panel derecho las combinaciones entre los SNPs, tanto de los datos observados y no observados por cada individuo. En este panel se comprueba el estado de la no monotonı́a de nuestros datos, ası́ como la variabilidad de missings en los diferentes locus. Gráficamente podemos también inspeccionar los Missing mediante pares de patrones Missing. Veámoslo a través de 4 ejemplos de combinaciones de SNPs de los 4 patrones anteriormente usados y mostrados en los gráficos de la figura 5.9. El área que contiene los puntos azules representa aquellas observaciones para los cuales ambos SNPs fueron observados. En el caso de los SNPs 645-294 podemos ver que hay 53 valores observados conjuntamente, tabla 5.9a, representado por las combinaciones de genotipos (genotipo=0, genotipo=0), (genotipo=0, genotipo=1), (genotipo=0, genotipo=2), (genotipo=1, genotipo=0), (genotipo=1, genotipo=1), (genotipo=2, genotipo=0), (genotipo=2, genotipo=1), (genotipo=2, genotipo=2). El área de los puntos rojos tanto en el sentido vertical como horizontal, son aquellas combinaciones entre valores missing y observados por SNP. 51 ● ● ● ● ● ● ● ● ● ● ● ● ● 2.0 ● ● ● ● ● ● ● ● ● 29 ● 12 29 ● ● ● ● ● ● 0.0 0.5 1.0 1.5 1.0 SNP417 0.5 0.0 1.0 0.5 0.0 SNP294 ● 1.5 ● 1.5 2.0 5.2. INSPECCIONANDO LOS MISSING DATA 22 ● 5 24 2.0 0.0 0.5 SNP645 1.5 2.0 2.0 (b) SNPs 297-417 ● ● ● ● ● ● ● ● ● 21 ● 3 21 ● ● ● ● ● ● 0.0 0.2 0.4 0.6 0.8 1.0 1.0 ● ● 0.5 0.0 SNP594 0.4 0.0 0.2 SNP197 0.6 1.5 0.8 1.0 (a) SNPs 645-294 ● 1.0 SNP297 ● ● 21 ● 6 21 ● 0.0 SNP510 ● 0.5 1.0 ● 1.5 2.0 SNP680 (c) SNPs 510-197 (d) SNPs 680-594 Figura 5.9: Plots Marginales El SNPs 645 presenta 29 Missing y el SNPs 294 tiene 29 Missing, también entre ellos existen 12 observaciones en que ambos SNPs coinciden siendo Missing. Los BoxPlots en azules resumen la distribución marginal de los SNPs correspondientes a los valores observados e igualmente los BoxPlos en rojo pero para los valores missing. Bajo MCAR, se espera que ambas distribuciones sean idénticas, es decir, las resumidas en los BoxPlots rojos y azules por cada SNP. Resumiendo los gráficos de la figura 5.9 se aprecian diferencias en los boxplots marginales, indicando que los marcadores no son MCAR respecto a otros marcadores. 52 5.3. CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Imputación Simple Esta metodologı́a descrita en el punto I de la sección 4.3.1 es válida bajo MCAR. Nos argumentamos para implementar este modelo debido a estos gráficos que a continuación exponemos que nos sugieren este problema. Ya habı́amos descartado la posibilidad de que los datos multivariados sean MCAR, sólo realizamos esta imputación como criterio extremista y además la teorı́a sugiere que este método introduce sesgos en el valor estimado y en su varianza. Para implementar este modelo procederemos directamente a imputar los Missing como heterocigotos, es decir, asignamos el mejor posible valor para los 5 SNP’s que seguidamente presentamos. Figura 5.10: SNP 645 Figura 5.11: SNP 294 Figura 5.12: SNP 9 Figura 5.13: SNP 194 53 5.3. IMPUTACIÓN SIMPLE SNP645 SNP294 SNP9 SNP194 SNP297 1 16 53 28 70 24 2 12 9 33 2 36 3 42 8 9 1 15 NA’s 29 29 29 26 24 Figura 5.14: SNP 297 En la figura 4.1 también veı́amos claramente la aparición de los missing en la frontera de los heterocigotos, al igual que en las figuras 5.6c y 5.6d. Aunque los SNPs 9 y 297 (Gráficos 5.12 y 5.14), no tienen bien definidos las diferentes clases de categorı́as, es decir, la clasificación de los homocigotos y heterocigotos están muy mezclados, los gráficos indican por sus posiciones pues una tendencia muy estrechas entre ellas, no como habı́amos visto para los demás SNPs, cuyas categorı́as están muy bien definidas, lo cual resultarı́a un problema según el supuesto que nos habı́amos planteado sobre la mala clasificación cuando se trataba de la categorı́a de los heterocigotos. Evidencia de sesgo en las imputaciones En la figura 5.15 podemos visualizar las diferencias entre los coeficientes de endogamia obtenidos por los casos observados y casos imputados de 10 SNPs, 5 de ellos visualizados en los gráficos anteriormente expuestos. Los SNPs marcados con los puntos rojos, son aquellos que resultaron significativos para la prueba χ2 de Pearson de los casos observados de los 10 SNPs que estamos analizando. Excepto por el SNP198 vemos la marcada distancia que existen entre los SNPs y la recta, indicando en general, la evidencia de sesgo resultante respecto a descartar missings, es decir, omitir aquellos casos no observados. Figura 5.15: Evidencia de sesgo entre la Imputación Simple y los Casos Observados 54 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS 5.4. Creando las imputaciones bajo MAR Imputaciones plausibles pueden dar razonables predicciones para los Missing Data y la variabilidad entre ellas debe reflejar un apropiado grado de incertidumbre. Rubin [10] recomienda que las imputaciones deban realizarse a través de un argumento bayesiano. Especificar un modelo paramétrico para los datos completos bajo MAR, asume una distribución a prior para los parámetros desconocidos del modelo y simula múltiples imputaciones independientes a partir de la distribución condicional de los Missing Data dado los valores observados por el teorema de Bayes. 5.4.1. Creando las imputaciones con MICE Mice es un paquete de R [19], que imputa datos multivariados incompletos vı́a Chained Equations, explicadas en la sección 4.4.2. A pesar de que previamente podemos tener conocimiento sobre los tipos de variables que analizamos ası́ como conocimientos sobre los criterios estadı́sticos fundamentales para la elección del método de imputación, es necesario realizar estudios a priori sobre los datos observados ası́ como analizar las relaciones entre las variables respuestas y covariables. 5.4.2. Selección de la matriz predictora Como requisito previo para la aplicación de los algoritmos de imputación que utilizan modelos de regresión, es necesario ajustar los modelos propuestos y verificar la significancia estadı́stica de los parámetros asociados a las covariables, es decir, son 2 pasos lo que envuelve la creación de las imputaciones, uno, la especificación del modelo de imputación, que es el paso más complejo en la imputación múltiple, ya que no siempre es conocido la distribución de las variables a imputar, y 2, la selección de los predictores que posiblemente sea el proceso más difı́cil. Existen criterios sobre la selección de dichos predictores. Veremos 2 metodologı́as. Criterio I Una estrategia es la comentada por [23] que consiste en 4 pasos a seguir, la cual enumeramos a continuación: 1. Incluir todas las variables que aparecen en el modelo de datos completos. De no hacerlo, puede sesgarse el análisis de datos completos, especialmente si el modelo de datos completos contiene fuertes relaciones predictivas. En particular esto significa que todos aquellos SNPs que se observaron completamente y todas las covariables son siempre parte del conjunto de predictores, por ejemplo las intensidades. 2. En adición, incluir los factores que son conocidos y que tienen influencias sobre la ocurrencia de los missing data (Estratificación, razones para la no-respuesta) deben 55 5.4. CREANDO LAS IMPUTACIONES BAJO MAR ser incluidas con motivos. Otras variables de interés son aquellas para las cuales las distribuciones difieren entre los grupos de respuestas y no respuestas. Estas pueden ser encontradas inspeccionando sus correlaciones respecto al indicador de respuesta de la variable con missings, es decir, la variable a ser imputada. Si la magnitud de esta correlación excede a cierto nivel, entonces la variable es incluida. 3. Incluir también aquellas variables que explican una considerable cantidad de varianza de la variable a imputar. Tales predictores ayudan a reducir la incertidumbre de las imputaciones. Ellas son crudamente identificadas por sus correlaciones con la variable a imputar. 4. Eliminar aquellas variables mencionadas en los 3 puntos anteriores que correspondan con las variables a imputar y que contengan muchos missing entre los subgrupos de casos incompletos. Un simple indicador es el porcentaje de casos observados dentro de este grupo, es decir, el porcentaje de casos utilizables. Criterio II. Análisis mediante regresión Como hemos descrito inicialmente, asumimos que estamos bajo el mecanismo MAR. Pero el hecho de asumir este mecanismo no es suficiente para continuar el proceso de imputación sin antes verificar que realmente admitir tal hipótesis se basa en algún fundamento. Realicemos la modelación de regresión logı́stica multinomial para un SNP observado completamente, incluyendo como predictores sus intensidades correspondiente. 0.8 0.6 Probability 0.0 0.2 Tabla 5.3: Regresión Logı́stica Multinomial BB AB AA 0.4 1:(intercept) 2:(intercept) 1:IA 2:IA 1:IB 2:IB 1.0 SNP 994 Estimate Std. Error t-value Pr(> |t|) 6.41 15538.70 0.00 1.00 2.01 22041.91 0.00 1.00 0.02 4.91 0.00 1.00 0.03 6.92 0.00 1.00 -0.01 5.42 -0.00 1.00 -0.04 7.99 -0.00 1.00 −2 0 2 4 Intensity A Figura 5.16: Regresión Logı́stica Multinomial Como se observa en la tabla 5.3 las intensidades no parecen ser predictores significativos para el SNP 994, cosa que nos ha extrañado mucho puesto que la obtención del genotipo es calculada por las medidas de las intensidades. La cuestión es que ocasionalmente cuando se ejecuta una regresión logı́stica podemos encontrarnos con el problema de la llamada separación completa o separación casi completa. Una separación completa 56 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS ocurre cuando la variable respuesta separa una variable de predicción o una combinación de variables predictoras completamente o viceversa. La separación completa o predicción perfecta puede ocurrir por varias razones, en nuestro caso, es debido a que la asignación del genotipo es dado según la cantidad de intensidad calculada por los 2 alelos medidos, por lo que esta está particionada por rangos, tal y como se explica en la sección 4.2. También podemos percatarnos en la tabla 5.3 que los errores estándar de los parámetros estimados son demasiado grandes, esto generalmente indica un problema de convergencia o algún grado de separación de datos [24]. Todo lo mentado anteriormente se puede corroborar con la figura 5.16 que expresa la relación entre el SNP 994 y su respectiva intensidad A, donde se describe que para baja intensidad de A, el genotipo resultante de mayor probabilidad es BB, para valores medios de A son los heterocigotos AB y para altas intensidades el genotipo AA. Viéndose claramente la separación de cada categorı́a respuesta. En las imputaciones se han incluido las intensidades a pesar de no ser significativas porque es evidente que guarda una relación casi determinista con la respuesta. Matriz Predictora La forma general de la matriz predictora quedarı́a de la siguiente manera como la de este ejemplo, las covariables Intensidades, son todas predictoras según su respectivo SNP y todos aquellos SNPs que fueron observados completamente, entiéndase que estos SNPs fueron seleccionados por la ubicación en la base de datos y no por su relación respecto a su posición fı́sica en el cromosoma. El conjunto de predictores serı́a X = [SN Pobs , IA, IB]. SNP645 SNP294 SNP9 SNP194 SNP297 SNP645 SNP294 SNP9 SNP194 SNP297 SNP645 0 0 0 0 0 A645 1 0 0 0 0 SNP294 0 0 0 0 0 A294 0 1 0 0 0 SNP9 0 0 0 0 0 A9 0 0 1 0 0 SNP194 0 0 0 0 0 A194 0 0 0 1 0 SNP297 0 0 0 0 0 A297 0 0 0 0 1 SNP8 1 1 1 1 1 B645 1 0 0 0 0 Tabla 5.4: Matriz Predictor SNP192 1 1 1 1 1 B294 0 1 0 0 0 SNP292 1 1 1 1 1 B9 0 0 1 0 0 SNP298 1 1 1 1 1 B194 0 0 0 1 0 SNP647 1 1 1 1 1 B297 0 0 0 0 1 5.4. CREANDO LAS IMPUTACIONES BAJO MAR 57 La matriz 5.4 debe analizarse de forma horizontal y nos indica aquellas variables que son predictoras de las otras o no, donde aquellas variables que no son predictoras están sujetas por los investigadores si se dejan en el modelo o no debido a su interés cientı́fico. En particular si analizamos el SNP 194 podemos ver que los SNPs 645, 294, 9 y 297 no son predictores para él, ni entre ellos y ası́ sucesivamente por cada SNP. Bajo el mecanismo MAR, la probabilidad de un patrón de no respuesta depende de los valores observados y sus covariables, es decir, p(R|Yobs , Z), por lo que se analizarán los modelos comentados en la sección 4.3, todos derivados de esta matriz predictora. M ICEY Y = p(R|Yobs ) ⇒ Modelo con predictores Yobs (5.1a) M ICEY Z = p(R|Z) ⇒ Modelo con predictores IA e IB (5.1b) M ICEY Y Z = p(R|Yobs , Z) ⇒ Modelo con predictores Yobs , IA e IB (5.1c) Entiéndase para el modelo 5.1a como aquel que sólo contiene SNPs como predictores, para el modelo 5.1b que sólo contiene las Intensidades de A y B del SNP correspondiente y para el modelo 5.1c ambos modelos anteriores combinados. 5.4.3. Chequeando el diagnóstico de los Missing Existen 2 técnicas de chequeo para valorar si nuestras imputaciones están correctas o al menos siguen una pauta adecuada en dependencia de la distribución de los datos observados. Una consta a través del chequeo del diagnóstico de las imputaciones y la otra mediante representaciones gráficas. 1 −1 0 1 2 3 4 5 6 7 8 9 10 0 1 2 3 4 5 6 7 8 9 10 0.5 0.5 2.0 0.0 1.5 0 1 2 3 4 5 6 7 8 9 10 0.0 −0.5 −0.5 0.0 −2 −1 −1 0.5 0 0 1.0 1 1 2 2 2.5 1.0 3 3 0 1 2 3 4 5 6 7 8 9 10 1.0 0 1 2 3 4 5 6 7 8 9 10 3.0 0 1 2 3 4 5 6 7 8 9 10 0 0.0 −1 −2 −2 0 1 2 3 4 5 6 7 8 9 10 −0.5 −1 0 0 0 0.5 2 1 1.0 1 2 1.5 2 4 2 3 2.0 Comprobar el diagnóstico de los datos imputados proporciona una manera de verificar la plausibilidad de las imputaciones, expresado de otra manera, el chequeo del diagnóstico debe ser un paso importante luego de la imputación, ya que verifica y evalúa si dichas imputaciones son plausibles. Un método de imputación mal seleccionado puede traer consigo malas imputaciones, veamos un ejemplo con un método de imputación para variables numéricas. La figura 5.17 demuestra el caSNP645 SNP294 SNP9 SNP194 SNP297 so tı́pico de selecionar un método de imputación no adecuado a los datos. Los puntos azules representan a los datos observados y los rojos son los imputados. Las imputaciones deben adquirir los valores que se podrı́an SNP417 SNP680 SNP594 SNP510 SNP197 haber obtenido si no se hubieran perdido, es decir, deben estar alrededor de los datos observados. Las imputaciones que son claramente imposibles, por ejemplo: recuentos negati0 1 2 3 4 5 6 7 8 9 10 0 1 2 3 4 5 6 7 8 9 10 0 1 2 3 4 5 6 7 8 9 10 Imputation number Figura 5.17: Método Regresión Lineal Bayesiana 58 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS vos, no deben ocurrir en los datos imputados. En general, las imputaciones deben respetar las relaciones entre las variables y reflejar la cantidad apropiada de la incertidumbre sobre sus verdaderos valores. En este ejemplo observamos que los puntos rojos fluctúan mucho entre los puntos azules, además los valores que asume llegan a ser negativos algunas veces; era de esperar debido a que elegimos un método de respuesta numérica. Verifiquemos lo anteriormente expuesto al SNP 645, por cada modelo planteado. Cada fila de las tablas 5.5, 5.6, 5.7 corresponden a una entrada missing en el SNP645, excepto las 3 últimas de cada tabla, que resumen la cantidad de veces que se imputó cada categorı́a en cada iteración. Podemos ver que las imputaciones creadas por los tres modelos son plausibles. Id 3 6 7 13 18 22 23 24 25 26 27 29 31 33 38 45 50 56 59 72 77 79 81 84 85 87 88 95 98 1 2 3 1 1 1 1 2 2 2 2 1 2 1 2 1 1 1 2 2 1 2 2 2 1 1 1 2 2 2 1 2 2 13 16 0 2 1 2 1 1 3 2 1 1 2 2 2 1 1 1 2 2 1 2 2 2 2 2 1 1 2 2 1 2 2 12 16 1 3 2 2 1 1 2 1 1 1 2 2 2 1 1 1 2 2 1 2 2 2 2 2 1 1 1 2 1 1 2 14 15 0 4 2 2 1 1 2 2 1 2 1 3 2 1 1 1 2 2 3 1 2 2 2 3 1 1 1 2 1 2 1 13 13 3 5 1 2 1 2 3 2 1 1 2 2 2 1 1 1 2 2 1 2 2 2 1 3 1 1 3 2 1 2 3 12 13 4 6 1 2 1 1 3 2 1 1 2 3 1 1 1 1 2 2 1 3 2 2 2 3 1 1 1 2 1 1 3 15 9 5 7 2 1 1 2 2 1 2 1 2 2 2 1 1 1 2 2 2 2 2 2 1 2 1 1 3 2 1 2 2 11 17 1 8 1 1 1 1 2 2 2 2 2 2 2 1 2 2 2 2 1 2 2 2 2 2 1 2 2 2 1 1 1 10 19 0 9 2 2 1 2 3 1 1 1 1 2 2 1 3 1 2 2 1 2 2 2 2 1 2 1 3 2 1 2 3 11 14 4 10 1 1 1 2 3 2 1 2 2 3 2 1 1 1 2 2 1 3 2 2 2 2 1 1 1 2 1 1 1 14 12 3 Tabla 5.5: MultiLogit M ICEY Y Z Id 3 6 7 13 18 22 23 24 25 26 27 29 31 33 38 45 50 56 59 72 77 79 81 84 85 87 88 95 98 1 2 3 1 3 2 3 3 1 3 3 2 3 3 3 3 2 3 2 1 1 1 2 3 1 2 3 2 1 1 3 3 2 7 8 14 2 3 3 3 2 2 3 3 3 1 3 3 2 2 3 2 3 1 1 3 2 2 2 1 3 2 2 3 3 3 4 10 15 3 3 1 3 3 3 3 2 3 1 3 2 1 1 3 1 3 3 1 3 2 3 1 3 3 3 2 3 1 3 8 4 17 4 1 2 3 2 1 2 3 3 3 3 2 1 2 3 2 3 3 1 3 1 2 3 1 1 1 2 3 3 3 8 8 13 5 3 2 3 2 3 3 3 1 3 2 3 3 1 3 1 3 3 2 1 1 3 3 3 3 1 3 3 3 3 6 4 19 6 3 3 3 3 1 2 3 1 3 1 3 3 3 3 3 3 3 3 3 3 3 3 3 3 2 3 3 3 3 3 2 24 7 3 3 3 3 3 2 1 3 3 3 3 3 3 1 3 3 3 3 3 2 1 1 3 3 2 3 3 3 1 5 3 21 8 2 2 3 2 3 3 1 1 3 3 3 1 1 3 3 2 3 3 1 1 3 1 3 2 3 1 2 3 3 8 6 15 9 3 3 3 3 3 2 1 1 2 3 3 3 3 3 3 3 3 2 1 3 3 3 3 3 3 2 1 3 3 4 4 21 Figura 5.18: Modelo M ICEY Y Z 10 2 2 3 1 3 1 1 1 3 3 3 3 3 3 2 3 3 1 1 1 3 1 3 1 1 3 3 3 3 10 3 16 Tabla 5.6: MultiLogit M ICEY Y Figura 5.19: Modelo M ICEY Y 59 5.4. CREANDO LAS IMPUTACIONES BAJO MAR Id 3 6 7 13 18 22 23 24 25 26 27 29 31 33 38 45 50 56 59 72 77 79 81 84 85 87 88 95 98 1 2 3 1 2 1 2 1 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 1 2 2 3 3 25 1 2 2 1 2 2 2 2 2 2 2 2 2 2 1 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 26 1 3 2 1 1 2 2 2 2 1 2 3 2 2 2 2 2 2 2 2 2 2 2 2 1 3 2 2 2 3 2 4 22 3 4 2 2 2 1 2 2 2 2 2 2 3 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 1 2 25 2 5 1 2 2 2 2 1 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 3 2 25 2 6 2 1 1 2 3 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 2 3 2 2 3 2 2 2 23 4 7 2 2 1 1 2 1 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 26 0 8 2 1 2 2 2 2 2 1 1 2 2 2 2 2 2 1 2 2 2 2 1 3 1 2 2 2 2 2 3 6 21 2 9 3 2 2 2 3 2 2 2 2 2 2 2 2 1 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 1 25 3 10 2 2 1 1 2 1 2 2 2 2 2 1 2 2 2 2 2 2 2 2 1 3 2 3 2 2 2 2 3 5 21 3 Tabla 5.7: MultiLogit M ICEY Z Figura 5.20: Modelo M ICEY Z Representando gráficamente las densidades tanto de los valores observados (curvas azules) e imputados (curvas rojas) de todas las variables se puede ver si las imputaciones son razonables (Gráficos 5.18, 5.19, 5.20). Diferencia significativa en las densidades entre los valores observados e imputados puede sugerir un problema que necesita ser revisado. Bajo MCAR, las distribuciones univariadas de los datos observados y los datos imputados se espera a que sean idénticos, sin embargo, bajo MAR ellos pueden ser diferentes, tanto en localización como en dispersión, pero su distribución multivariada se supone que es idéntica. Podemos observar en las tablas 5.5, 5.6 y 5.7 las últimas 3 filas de cada una de ellas, que están en correspondencias con lo que se visualiza en los gráficos 5.18, 5.19 y 5.20, es decir, en los modelos donde se incluyeron las intensidades hubo más tendencia a imputar heterocigotos (Modelos M ICEY Y Z y M ICEY Z ), sin embargo en el modelo donde sólo se incluyen los SNPs observados pues las densidades siguen la misma pauta de los datos observados (Modelo M ICEY Y ), es decir, imputó sobre las tres categorı́as y siempre predominando el genotipo BB. Respecto a los modelos M ICEY Y Z y M ICEY Z , podemos decir que el modelo M ICEY Y Z , tuvo cierta inclinación a equilibrar aquellas categorı́as de menos valores observados, llegando a igualar los genotipos AA y AB, todo lo contrario al modelo M ICEY Z que sus mayor propensión fue imputar heterocigotos, este modelo refleja lo comentado en la sección 5.3, donde veı́amos gráficamente el problema de la generación de Missing en las fronteras entre los homocigotos y heterocigotos. Todo lo descrito podemos observarlo en las tablas 5.8, 5.9 y 5.10. 60 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Imp 1 2 3 4 5 6 7 8 9 10 AA 29 28 30 29 28 31 27 26 27 30 AB 28 28 27 25 25 21 29 31 26 24 BB 42 43 42 45 46 47 43 42 46 45 Tabla 5.8: M ICEY Y Z 5.4.4. Imp 1 2 3 4 5 6 7 8 9 10 AA 23 20 24 24 22 19 21 24 20 26 AB 20 22 16 20 16 14 15 18 16 15 Tabla 5.9: M ICEY Y BB 56 57 59 55 61 66 63 57 63 58 Imp 1 2 3 4 5 6 7 8 9 10 AA 19 18 20 18 18 18 19 22 17 21 BB 43 43 45 44 44 46 42 44 45 45 Tabla 5.10: M ICEY Z Evidencia de sesgos en las imputaciones bajo MICE (a) M ICEY Y Z AB 37 38 34 37 37 35 38 33 37 33 (b) M ICEY Y (c) M ICEY Z Figura 5.21: Evidencia de sesgo en las imputaciones realizadas al SNP645 61 5.4. CREANDO LAS IMPUTACIONES BAJO MAR Schafer (1999) [25], da respuesta algunas interrogantes que surgen sobre el procedimiento Imputación Múltiple, dentro de estas, se encuentra el hecho de eliminar aquellos casos no observados. Nosotros hemos analizado los sesgos que se generan en el proceso de inferencia cuando la falta de respuesta es importante y comparado con las imputaciones realizadas por los distintos modelos, como mismo se hizo en la sección 5.3 bajo el método de Imputación Simple. En los gráficos de la figura 5.21 podemos observar la evidencia de sesgo resultante de las imputaciones realizadas por cada método respecto a descartar los missings. En el modelo que menos sesgo se observa fue el M ICEY Y donde sus predictores fueron aquellos SNPs observados completamente, dado en el gráfico 5.21b. En términos del coeficiente de endogamia, podemos observar en cada gráfico el pooling obtenido de la combinación de las 10 imputaciones realizadas. En los modelos M ICEY Y Z y M ICEY Z el coeficiente baja respecto al coeficiente de los casos observados, que es lo mismo decir que se imputan relativamente más heterocigotos. (a) M ICEY Y Z (b) M ICEY Y (c) M ICEY Z Figura 5.22: Evidencia de sesgo en las imputaciones para diferentes SNPs 62 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Si analizamos todos los SNPs en conjunto, podemos ver que el modelo M ICEY Z (gráfico 5.22a) tuvo el mismo comportamiento para todos ellos. Todos se encuentran por debajo de la recta y = x indicando la ausencia de heterocigotos. El modelo M ICEY Y Z (gráfico 5.22c) tiene un comportamiento similar excepto por el SNP194 que presentó una sobreimputación de heterocigotos y el modelo M ICEY Y (gráfico 5.22b) como ya habı́amos comentado tuvo la tendencia de crear imputaciones muy similares a la de los datos completos, por esto vemos que casi todos SNPs coinciden sobre la recta. En términos de sesgo podemos decir que, el modelo M ICEY Z fue el que más mostró evidencia de sesgo resultante de las imputaciones realizadas respecto a descartar los missings, indicando de manera general para todos los SNPs analizados que el hecho de imputarlos pues influye en la inferencia estimada del coeficiente de endogamia. 5.4.5. Creando las imputaciones con CAT El paquete ’CAT’ [26], realiza análisis de variables categóricas con valores Missing, sus métodos de imputación están basados en los descritos por Joseph L. Schafer [15]. Existen diversas metodologı́as para el trato de Datos Categóricos Multivariantes Incompletos. Aplicaremos 2 técnicas combinadas entre sı́, el Algoritmo EM y Algoritmo DA explicados en la sección 4.4.3. Los pasos a seguir son: a. Crear por cada SNPs un patrón monótono, por ende, se implementó el proceso de imputación SNPs por SNPs, convirtiéndose en un mecanismo univariado. b. Se aplicó el algoritmo EM para encontrar los estimadores máximos verosı́miles de las probabilidades bajo el modelo multinomial saturado. c. Se implementó el algoritmo DA a partir de las probabilidades estimadas en (b). d. Se desarrollan imputaciones aleatorias simples de los Missing Data usando los estimadores encontrados en (c). e. Se repiten los pasos (c)-(d) hasta obtener m-imputaciones. En nuestro caso, usaremos el método de DA monótono (MDA), es decir, como imputaremos SNP a SNP pues al convertirse en patrones monótonos el MDA tiende a converger más rápidamente que el método DA. Este procedimiento es lo que conocemos como “Aplicar iterativamente métodos para patrones monótonos”, creado por Tanner y Wong (1987) y Li (1985) cada uno con objetivos diferentes y que comentamos en la sección 4.4.3. 63 5.4. CREANDO LAS IMPUTACIONES BAJO MAR 5.4.6. Chequeando el diagnóstico de los Missing En términos de probabilidades podemos plantear el modelo Imp 1 2 3 4 5 6 7 8 9 10 AA 26 21 24 24 20 22 21 22 20 21 AB 18 17 17 15 19 13 15 15 17 15 BB 55 61 58 60 60 64 63 62 62 63 CATY Y = p(R|Yobs ) (5.2a) La idea básica es, mediante el método EM encontrar los estimadores máximos verosı́miles de los parámetros y con estos elaborar las imputaciones al azar en virtud de su distribución predictiva dados los datos observados y el valor actual de θ luego mediante el método de simulación de Monte Tabla 5.11: CATY Y Carlo vı́a Cadenas de Markov encontrar la distribución posteriori de los parámetros y ası́ hasta la cantidad de veces que se desea imputar. En la tabla 5.11 observamos que la categorı́a que menos se imputó fue la de heterocigotos. Dentro de cada genotipo por cada imputación se observa similitud en cuanto al número de imputación. 5.4.7. Evidencia de sesgo en las imputaciones bajo CAT (a) CATY Y para el SNP645 (b) Pooling CATY Y Figura 5.23: Evidencia de sesgo en las imputaciones para diferentes SNPs Observando el gráfico 5.23a notamos que los valores del coeficiente de endogamia imputados son aproximadamente cercanos al valor del coeficiente obtenido descartando los datos no observados. Dicha conclusión también se extrapola al pooling realizado a todos los SNPs analizados que aparecen en el gráfico 5.23b. 64 5.4.8. CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Creando las imputaciones con MIX El paquete ’MIX’ [27], es un soft que realiza estimación e imputación múltiple de datos mixtos con variables categóricas y continuas. Nosotros aplicaremos 2 combinaciones de técnicas como en la sección 5.4.5. Utilizaremos los métodos EM y DA para modelos de localización general sin restricciones. Dichos métodos se basan en el marco de Schafer [15] y Rubin [11]. Los pasos a seguir son parecidos a los mencionados en la sección 5.4.5, la diferencia es que junto a la variable a imputar se incluyen todas aquellas variables continuas que pueden inferir en la imputación: a. Se aplicó el algoritmo EM para encontrar los estimadores máximos verosı́miles de las variables a imputar. b. Se implementó el algoritmo DA a partir de las probabilidades estimadas en (a). c. Se desarrolla imputaciones aleatorias simples de los Missing Data usando los estimadores encontrados en (b). d. Se repiten los pasos (b)-(c) hasta m-imputaciones. La idea básica de este procedimiento consiste en aplicar el método de Monte Carlo vı́a Cadena de Markov para generar los valores a posteriori de los parámetros del modelo de localización general sin restricción. Inicialmente en cada paso, los Missing Data son aleatoriamente imputados, primero encontrando los parámetros con el método EM y unos nuevos valores de los parámetros son buscados mediante el método DA, es decir, estos a través de la distribución a posteriori dado los valores completos. 5.4.9. Chequeando el diagnóstico de los Missing Imp 1 2 3 4 5 6 7 8 9 10 AA 16 16 16 17 16 16 16 16 16 16 AB 40 40 40 38 40 41 39 40 41 35 BB 43 43 43 44 43 42 44 43 42 48 Se nota que el sistema no imputó sobre las categorı́a 1 y 3 en casi ninguna iteración. Las imputaciones intra genotipos son muy similares, también existe mucha similaridad entre los genotipos BB y AB. Véase tabla 5.12. Tabla 5.12: M IXY Z El modelo a plantear mediante este sistema de imputación lo llamaremos M IXY Z = p(R|Z), donde ya habı́amos realizado una modelación similar pero basado en el soft MICE. Comparémosla a ver la diferencia entre los sistemas de imputación. 65 5.4. CREANDO LAS IMPUTACIONES BAJO MAR Imp 1 2 3 4 5 6 7 8 9 10 AA 16 16 16 17 16 16 16 16 16 16 AB 40 40 40 38 40 41 39 40 41 35 BB 43 43 43 44 43 42 44 43 42 48 Tabla 5.13: M IXY Z Imp 1 2 3 4 5 6 7 8 9 10 AA 19 18 20 18 18 18 19 22 17 21 AB 37 38 34 37 37 35 38 33 37 33 BB 43 43 45 44 44 46 42 44 45 45 SNP 645 AA AB BB NA 16 12 42 29 Tabla 5.15: Descriptiva Tabla 5.14: M ICEY Z Podemos observar que los 2 métodos imputan muy similarmente, uno a través de Chained equation (MICE) y el otro a través de la combinación de los métodos EM y AD (MIX). En uno se declara inicialmente los predictores y se imputa usando el modelo multinomial saturado y el otro a través del modelo de localización general, respectivamente. También podemos comentar que ambas metodologı́as crean las imputaciones corroborando lo que habı́amos comentado en la imputación simple, donde casi todos los missing se imputaron en la categorı́a de los heterocigotos. 5.4.10. Evidencia de sesgo en las imputaciones bajo MIX (a) M IXY Y para el SNP645 (b) Pooling M IXY Y Figura 5.24: Evidencia de sesgo en las imputaciones para diferentes SNPs En la figura 5.24 podemos observar que la inclusión de las intensidades conduce a estimaciones más bajas del coeficiente de endogamia. Esta conclusión la podemos extender a todos los SNPs analizados, como se observa en el gráfico 5.24b. Este modelo presentó el mismo comportamiento que el modelo M ICEY Z . 66 5.4.11. CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Comparando las Imputaciones Sigamos usando el mismo ejemplo basado en el SNP 645, hagamos una sencilla comparación entre las imputaciones por los diversos modelos planteados. Imp 1 2 3 4 5 6 7 8 9 10 AA 29 28 30 29 28 31 27 26 27 30 AB 28 28 27 25 25 21 29 31 26 24 BB 42 43 42 45 46 47 43 42 46 45 Imp 1 2 3 4 5 6 7 8 9 10 Tabla 5.16: M ICEY Y Z Imp 1 2 3 4 5 6 7 8 9 10 Tabla 5.19: CATY Y AA 23 20 24 24 22 19 21 24 20 26 AB 20 22 16 20 16 14 15 18 16 15 BB 56 57 59 55 61 66 63 57 63 58 Imp 1 2 3 4 5 6 7 8 9 10 Tabla 5.17: M ICEY Y AA 26 21 24 24 20 22 21 22 20 21 AB 18 17 17 15 19 13 15 15 17 15 BB 55 61 58 60 60 64 63 62 62 63 AA 19 18 20 18 18 18 19 22 17 21 AB 37 38 34 37 37 35 38 33 37 33 BB 43 43 45 44 44 46 42 44 45 45 Tabla 5.18: M ICEY Z Imp 1 2 3 4 5 6 7 8 9 10 AA 16 16 16 17 16 16 16 16 16 16 AB 40 40 40 38 40 41 39 40 41 35 BB 43 43 43 44 43 42 44 43 42 48 Tabla 5.20: M IXY Z Como habı́amos comentado las imputaciones realizadas por M ICEY Z y M IXY Z están más en correspondecia con la tabla 5.15, ya que hubo pocas imputaciones sobre los homocigotos indicando esto el problema al que hacı́amos referencia sobre la asignación de genotipado en la frontera de los heterocigotos. Ambos modelos están condicionados por las intensidades de el mismo SNP, por lo que esto contrasta el hecho de que si sólo utilizamos las intensidades, se imputan más heterocigotos a que si incluimos en los modelos las intensidades y otros SNPs. También podemos cerciorarnos que M IXY Z imputa más heterocigotos que M ICEY Z El modelo CATY Y no hizo casi imputaciones sobre los heterocigotos, repartiendo todos los Missing en las categorı́as de los homocigotos, todo lo contrario a lo comentado en la sección 5.3. Por otro lado el modelo M ICEY Y , es un modelo multivariado que usa como predictores para las imputaciones a otros SNPs observados completamente. Este modelo aunque realizó más imputaciones sobre los heterocigotos tuvo la misma tendencia que el modelo CATY Y , aumentar el número de homocigotos. Esto pudiera ser un indicador de que aquellos missing que se encuentran entre las fronteras de homocigotos y heterocigotos pues tienen más posibilidades de ser homocigotos. Visto con otro enfo- 67 5.5. POOLING que pudiéramos decir que las imputaciones basadas desde el punto de vista multivariado como univariado tienden a ser las mismas, indicando que el hecho de incluir o no otros SNPs al modelo tienen el mismo efecto. Por lo contrario el modelo M ICEY Y Z imputó casi todos los Missing generalmente en los genotipos AA y AB, cuyas imputaciones entre estos 2 genotipos son casi iguales entre ellas, además fue muy conservativo en las imputaciones intra categorı́as. La tendencia de este modelo fue equilibrar las categorı́as de menos conteo. Habı́amos hecho referencia en la sección III, al modelo que describe que el porcentaje de significativos no tiene porque ser exactamente igual con la intensidad de A y con la intensidad de B y justamente es lo que reflejan las imputaciones basadas en M ICEY Y Z , donde hubo más tendencia a imputar aquellos Missing relacionados con la Intensidad A. 5.5. Pooling Nuestra variable de interés es el coeficiente de endogamia y a través de él realizaremos los test para el HWE. fˆcc fˆis fˆ std.err df p value CI Inf CI Sup rM γ Inferencia Múltiple Imputación SNP645 SNP294 SNP9 SNP194 SNP297 0.60 0.56 -0.018 0.49 0.026 0.11 0.032 -0.30 -0.1 -0.22 0.45 0.11 91.18 0.00 0.24 0.67 0.46 0.33 0.44 -0.02 0.12 0.11 135.45 278.20 0.00 0.87 0.19 -0.24 0.68 0.20 0.35 0.22 0.27 0.19 0.61 0.11 48.40 0.00 0.38 0.84 0.76 0.45 -0.02 0.11 329.48 0.86 -0.24 0.20 0.20 0.17 fˆcc fˆis fˆ std.err df p value CI Inf CI Sup rM γ Tabla 5.21: Modelo M ICEY Y Z fˆcc fˆis fˆ std.err df p value CI Inf CI Sup rM γ 0.28 -0.04 0.13 0.11 200.99 399.22 0.03 0.72 0.03 -0.25 0.53 0.17 0.27 0.18 0.22 0.15 0.45 0.16 23.83 0.01 0.12 0.77 1.59 0.64 Tabla 5.23: Modelo M ICEY Z 0.57 0.10 147.62 0.00 0.38 0.76 0.33 0.26 0.59 -0.05 0.24 0.10 3256.12 1913.06 0.01 0.63 0.12 -0.25 1.07 0.15 0.06 0.07 0.05 0.07 0.49 0.17 26.10 0.01 0.15 0.84 1.42 0.62 0.02 0.11 1325.67 0.87 -0.19 0.22 0.09 0.08 Tabla 5.22: Modelo M ICEY Y Inferencia Múltiple Imputación SNP645 SNP294 SNP9 SNP194 SNP297 0.60 0.56 -0.018 0.49 0.026 0.11 0.032 -0.30 -0.1 -0.22 0.25 0.10 617.03 0.02 0.05 0.46 0.14 0.12 Inferencia Múltiple Imputación SNP645 SNP294 SNP9 SNP194 SNP297 0.60 0.56 -0.018 0.49 0.026 0.11 0.032 -0.30 -0.1 -0.22 -0.05 0.11 655.04 0.64 -0.26 0.16 0.13 0.12 fˆcc fˆis fˆ std.err df p value CI Inf CI Sup rM γ Inferencia Múltiple Imputación SNP645 SNP294 SNP9 SNP194 SNP297 0.60 0.56 -0.018 0.49 0.026 0.11 0.032 -0.30 -0.1 -0.22 0.62 0.10 276.63 0.00 0.42 0.81 0.22 0.19 0.55 0.14 48.58 0.00 0.27 0.82 0.76 0.45 -0.01 0.13 65.11 0.96 -0.26 0.25 0.59 0.39 0.47 0.25 368.44 0.06 -0.03 0.97 0.19 0.16 Tabla 5.24: Modelo CATY Y 0.00 0.12 80.18 0.06 -0.24 0.25 0.50 0.35 68 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS fˆcc fˆis Inferencia Múltiple Imputación SNP645 SNP294 SNP9 SNP194 SNP297 0.60 0.56 -0.018 0.49 0.026 0.11 0.032 -0.30 -0.1 -0.22 fˆ 0.13 std.err 0.10 df 18261.05 p value 0.22 CI Inf -0.08 CI Sup 0.33 rM 0.02 γ 0.02 0.13 -0.12 0.11 0.10 1286.94 675.58 0.22 0.24 -0.08 -0.33 0.35 0.08 0.09 0.13 0.09 0.12 0.13 0.15 39.25 0.40 -0.18 0.44 0.92 0.50 -0.08 0.10 7011.61 0.40 -0.28 0.12 0.04 0.04 Tabla 5.25: Modelo M IXY Z Figura 5.25: Posiciones relativas para los estimados de fˆ. cc: Casos Observados. is: Imputación Simple. m1: M ICEY Y Z . m2: M ICEY Y . m3: M ICEY Z . m4: CATY Y . m5: M IXY Z Las componentes que vemos en cada tabla, son las descritas en Rubin [10]: - fˆcc es el estimador del coeficiente de endogamia de los casos observados por cada SNP. - fˆis es el estimador del coeficiente de endogamia de la imputación simple. - fˆ es el promedio del estimador del coeficiente de endogamia de las m-imputaciones. - std.err es el error estándar incorporando ambas varianzas de fˆ, la varianza intra imputación y entre imputación. Que no es más que la raı́z cuadrada de la varianza total. - df son los grados de libertad asociados con la distribución tStudent . - pV alue de dos colas para H0 : f = 0 - CIInf y CISup son los intervalos de confianza de fˆ del (100 ∗ (1 − α)) % - rM : es el incremento relativo en varianza debido a la no respuesta. - γ: es la fracción estimada de la información Missing. 5.5. POOLING 69 La figura 5.25 muestra la posición relativa de los estimadores de fˆ para los distintos modelos que se exponen. En ellos podemos ver las similitudes de los distintos modelos. (1) Los estimadores de los casos observados (cc), los modelos m2 y m4 coinciden en todos los SNPs analizados en cuanto a sus cercanı́as, (2) los modelos ImpSim (is, caso más extremista) y m5 en todos los SNPs tuvieron el comportamiento más cercano a 0 y siempre en el extremo izquierdo. (3) Los modelos m1 y m3 no tienen ningún patrón definido, aunque m3 suele estar por debajo de m1. Esto resume lo visto en las imputaciones que se comentaron en la sección 5.4.11. El modelo m5 representarı́a el caso que lleva las estimaciones más cercanas al HWE. Según los valores de fˆ y los pV alues asociados podemos decir que aceptamos que estos SNPs están bajo HWE. Si analizamos en conjunto los estimadores de los SNPs podemos observar que se formaron 2 grupos en cuanto al valor estimado del coeficiente de endogamia, un grupo compuesto por los SNPs: SNP645, SNP294 y SNP194, y el otro por el resto. Esto puede deberse a lo comentado en la sección 5.3, constatando que la eficacia de los procedimiento depende de la variable de análisis, de la tasa de no respuesta y de su distribución en la muestra y permite afirmar que si una técnica de imputación resultó adecuada para una variable, no significa que su uso se debe generalizar sin analizar las condiciones en que se generó la falta de respuesta en otras variables de interés [14]. Concretando lo que queremos decir, podemos ver que en todos los métodos que aplicamos, los SNPs 9 y 297 tuvieron un comportamiento muy similar, donde los coeficientes de endogamia estimados están muy alrededor del valor cero. El mismo comportamiento en cuanto a cercanı́as de los modelos y las posiciones de sus valores estimados del coeficiente de endogamia, la tuvieron los SNPs 645 y 294. El SNP194 tuvo un patrón de posición más similar a estos últimos pero con ciertas diferencias a los valores tomados por el parámetro de interés, cuya peculiaridad es que según el gráficos 5.13, las categorı́as de homocigotos están muy desequilibradas ya que en el genotipo AA sólo existe un valor clasificado. En términos de los valores obtenidos en las tablas 5.21 -5.25 iniciaremos explicando que tomamos M = 10, cuya justificación comentamos en la sección 4.4.1 de que el hecho de usar tantas iteraciones, es debido a que nuestras variables, algunas tienen un alto porcentaje de Missing. Schafer [15] declara que en aplicaciones, los cálculos de rM y γ son altamente recomendados ya que son muy interesantes y útiles en el diagnóstico para la evaluación de 70 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS cómo los datos faltantes contribuyen a la incertidumbre inferencial sobre f . Referente a rM que como habı́amos escrito anteriormente, es el incremento relativo en varianza debido a la no respuesta, llamada ası́ porque W̄ (ecuación 4.2) representa la varianza total estimada cuando no hay información missing sobre f , es decir, cuando B = 0 (ecuación 4.3), para M grande y/o rM pequeño, los grados de libertad serán grandes y la ecuación 4.4.1 será aproximadamente normal. Este elemento es de mucha utilidad ya que nos indicarı́a que las diferentes categorı́as en cada SNP están idénticamente distribuidas y son estadı́sticamente independientes [15]. Podemos observar que excepto en el SNP194; los demás SNPs, para los modelos M IXY Z y M ICEY Z en este orden, presentan grados de libertad altamente grandes y rM relativamente pequeños. En términos de eficiencia, en Rubin [10, p.114] se muestra que la eficiencia de un γ −1 estimador en m-imputaciones es aproximadamente (1 + M ) , donde γ es la fracción estimada de la información Missing, dicha fracción cuantifica cuánto más precisa es la estimación que podrı́a haber sido si los datos no hubieran sido Missing. Si nos remitimos a las tablas 5.21 -5.25 planteadas, podemos observar que aquellos modelos cuyos estimadores que más eficiencia alcanzaron fueron, en primer lugar, M IXY Z y en segundo M ICEY Z excepto por el SNP194, con una eficiencia relativa entre un 98-99 % para 10 imputaciones, véase tabla 4.1. 5.6. Creando las imputaciones bajo MNAR. Análisis de sensibilidad En la sección 4.3.1 hicimos referencias a una series de modelos que podı́amos plantear para marcadores genéticos. En esta sección analizaremos el descrito en el punto IV de la sección 4.3.1. De este modelo podemos derivar 2 modelos más, uno al cual lo notaremos como M N AR1 , como contraparte del modelo M ICEY Y Z (ecuación 5.1a) y el otro, M AN R2 , como contraparte del modelo M ICEY Y (ecuación 5.1c). Los modelos a los que hacemos referencia lo implementaremos a través del soft MICE. Ya habı́amos explicado que el Soft MICE trabaja con el sistema de Chained Equations, cuya primera definición es la matriz predictora e hicimos referencia que esta trabaja con 2 matrices de correlaciones. En general el procedimiento calcula por cada par de variables, 2 tipos de correlaciones, usando todos los casos válidos por pares. La primera correlación usa los valores de la variable respuesta y los predictores. La segunda correlación usa el indicador de respuesta (R) de la variable respuesta y los valores predictores. Si el valor de estas correlaciones (en valor absoluto) superan el punto umbral declarado por el investigador, 5.6. CREANDO LAS IMPUTACIONES BAJO MNAR. ANÁLISIS DE SENSIBILIDAD 71 entonces los predictores serán incluidos para el proceso de imputación. En adición el procedimiento elimina los predictores el cual la proporción de casos usables no cumple con el mı́mimo especificado que por lo general es el 50 %. Variables r(SNP645) r(SNP294) r(SNP9) r(SNP194) r(SNP297) r(R645 ) SNPs: SNPs Incompletos SNP645 1.00 0.05 -0.03 -0.07 -0.14 SNP294 0.05 1.00 -0.13 -0.11 0.08 -0.144 SNP9 -0.03 -0.13 1.00 0.19 0.06 -0.007 SNP194 -0.07 -0.11 0.19 1.00 -0.04 -0.091 SNP297 -0.14 0.08 0.06 -0.04 1.00 -0.153 SNPs: SNPs Completos SNP8 0.12 -0.10 -0.10 -0.06 0.01 SNP192 0.16 0.00 -0.06 0.06 -0.00 SNP292 -0.20 -0.06 -0.02 0.02 SNP298 -0.04 -0.10 0.04 0.05 0.03 SNP647 -0.03 -0.23 0.04 -0.11 0.05 Covariables IA645 0.89 0.11 -0.01 -0.08 -0.06 IB645 -0.82 0.02 -0.00 -0.01 0.18 IA294 0.05 0.92 -0.30 -0.08 0.05 IB294 0.11 -0.72 0.08 0.07 -0.01 IA9 0.16 -0.00 0.52 0.05 -0.09 IB9 0.16 0.11 -0.78 -0.08 -0.02 IA194 0.05 -0.06 0.08 0.96 0.04 IB194 0.20 0.06 0.06 -0.23 -0.02 IA297 0.23 0.18 -0.05 0.03 0.57 IB297 0.30 0.01 -0.05 0.04 -0.85 Tabla 5.26: Resumen de las variables que son usadas para la imputación. Las columnas de la 2-6 contiene las correlaciones de las variables filas respecto a los SNPs Missing. Columna 7 es un ejemplo de la correlación entre el indicador de respuesta y los datos del SNP 645. Columna 8 es el porcentaje de casos usables que es igual al porcentaje de los datos observados de las variables filas entre el subgrupo de casos que tienen Missing para el SNP 645 En la tabla 5.26 reflejamos lo descrito anteriormente. Las 2 últimas columnas son referidas al ejemplo con el SNP 645 que es el que ilustraremos, pero para cada SNP con % 58.6 65.5 72.4 79.3 72 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Missing se debe realizar el mismo proceso. Los SNPs con Missing serán predictores si cumplen simultáneamente tener en valor absoluto un r(RSN P ) > 0,1 y más del 50 % de casos utilizables y para las intensidades pues serán predictores para sus correspondientes SNPs. Seguidamente expondremos cómo quedarı́a la matriz predictora para el caso de los 5 SNPs. SNP645 SNP294 SNP9 SNP194 SNP297 SNP645 SNP294 SNP9 SNP194 SNP297 SNP645 0 0 1 1 1 A645 1 0 0 0 0 SNP294 1 0 1 1 0 A294 0 1 0 0 0 SNP9 0 1 0 1 1 A9 0 0 1 0 0 SNP194 0 1 1 0 1 A194 0 0 0 1 0 SNP297 1 0 1 0 0 A297 0 0 0 0 1 SNP8 1 1 1 1 1 B645 1 0 0 0 0 SNP192 1 1 1 1 1 B294 0 1 0 0 0 SNP292 1 1 1 1 1 B9 0 0 1 0 0 SNP298 1 1 1 1 1 B194 0 0 0 1 0 SNP647 1 1 1 1 1 B297 0 0 0 0 1 Tabla 5.27: Matriz Predictora: MNAR 5.6.1. Chequeando el diagnóstico de los Missing Imp 1 2 3 4 5 6 7 8 9 10 AA 35 31 36 30 32 36 34 35 29 34 AB 21 25 20 26 22 21 21 19 27 21 BB 43 43 43 43 45 42 44 45 43 44 fˆ std.err df p value CI Inf CI Sup rM γ Tabla 5.29: M N AR1 Tabla 5.28: M N AR1 Imp 1 2 3 4 5 6 7 8 9 10 AA 28 24 26 26 21 27 26 21 27 25 AB 16 19 13 19 16 16 13 19 16 16 BB 55 56 60 54 62 56 60 59 56 58 Tabla 5.30: M N AR2 SNP645 SNP294 SNP9 SNP194 SNP297 0.54 0.43 0.01 0.59 0.05 0.10 0.11 0.11 0.11 0.11 80.03 1408.79 1275.06 69.94 705.35 0.00 0.00 0.94 0.00 0.67 0.34 0.21 -0.20 0.37 -0.16 0.75 0.66 0.21 0.81 0.26 0.50 0.09 0.09 0.56 0.13 0.35 0.08 0.09 0.38 0.12 fˆ std.err df p value CI Inf CI Sup rM γ SNP645 SNP294 SNP9 SNP194 SNP297 0.63 0.49 -0.05 0.60 0.03 0.10 0.21 0.11 0.19 0.11 87.22 717.03 230.16 16.19 368.69 0.00 0.02 0.66 0.01 0.82 0.44 0.09 -0.27 0.19 -0.19 0.82 0.90 0.17 1.02 0.24 0.47 0.13 0.25 2.93 0.19 0.34 0.11 0.20 0.77 0.16 Tabla 5.31: M N AR2 5.6. CREANDO LAS IMPUTACIONES BAJO MNAR. ANÁLISIS DE SENSIBILIDAD 73 Si proyectamos los valores de estos nuevos modelos en el gráfico 5.25 observaremos el comportamiento de estos respecto a los demás planteados bajo el mecanismo MAR. Figura 5.26: Posiciones relativas para los estimados de fˆ. cc: Casos Observados. is: Imputación Simple. m1: M ICEY Y Z . m2: M ICEY Y . m3: M ICEY Z . m4: CATY Y . m5: M IXY Z . m6: M N AR1 . m7: M N AR2 La figura 5.26 muestra la posición relativa de los estimadores de fˆ para los distintos modelos que se exponen como mismo vimos en la figura 5.25. En ellos podemos ver las similitudes de los nuevos modelos M N AR0 s. Excepto en el SNP645, en el resto podemos observar la asociación que tienen con el modelo M ICEY Y Z , cosa que parece contradictoria pues el modelo M N AR2 es la contraparte del modelo M ICEY Y y se esperaba que el estimador estuviera más cercano a este, sin embargo no resultó ası́ en todos los SNPs. Esto nos indica que el hecho de incluir los SNPs con Missings pues tiene el mismo efecto que si analizamos el modelo sin incluirlos, es decir, el parámetro estimado bajo MNAR como MAR son muy aproximados. En términos de los errores estándares, al SNP645 SNP294 SNP9 SNP194 SNP297 secc 0.10 0.13 0.12 0.32 0.16 comparar los valores de estos, generados seis 0.10 0.10 0.09 0.07 0.10 por los diversos métodos de imputación a seMICEY Y Z 0.11 0.12 0.11 0.11 0.11 cada SNPs, se podrı́a argumentar que toseMICEY Y 0.10 0.24 0.10 0.17 0.11 dos los métodos generan un error estándar seMICEY Z 0.10 0.13 0.11 0.16 0.11 seCATY Y 0.10 0.14 0.13 0.25 0.12 similar por cada SNPs y si nos dejáramos seMIXY Z 0.10 0.11 0.10 0.15 0.10 llevar por esta simple conclusión pudiéraseMNAR1 0.10 0.11 0.11 0.11 0.11 mos decir que cualquiera de estos métodos seMNAR2 0.10 0.21 0.11 0.19 0.11 se pudiera utilizar para imputar, sim embarTabla 5.32: Errores Estándares go se debe analizar más a fondo con respecto a la forma de imputar cada uno de ellos. 74 5.7. CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS Comparación de modelos de imputación respecto a HWE Por cada SNPs se especificaron un conjunto de métodos de imputación y se realizaron los pooling por cada uno de estos usando el parámetro del coeficiente de endogamia y a través de la metodologı́a aplicada por Rubin [10], además se llegó a que el mejor modelo y más eficiente fue el M IXY Z (sección 5.5). Existen diversas técnicas para la realización del pooling, muy similar a la explicada por Rubin, estas constan a través de la combinación de estadı́sticos la cual explicaremos seguidamente y tomamos como referencia [28, p.26], [15, p.115]. Conocemos que el coeficiente f se puede expresar por la ecuación 2.20, que es equivalente si planteamos la expresión: Z = sign(fˆ) · p χ2 (5.3) Es decir, supongamos que tenemos k-estadı́sticos X 2 , uno por cada conjunto de datos con k-ésima imputaciones múltiples; podemos calcular k-estadı́sticos Z como en la ecuación 5.3 que bajo la hipótesis nula cada Zi :N (0, 1). Si tomamos Z̄ 2 y B (ecuación Z̄ 2 4.3) como la varianza entre las Z 0 s entonces bajo la hipótesis nula el estadı́stico 1+B Z tiene aproximadamente una χ21 con lo que podemos calcular el pvalue y comprobar si se cumple la hipótesis de que nuestro parámetro f = 0. fˆ fˆM ICEY Y Z Parámetros Z se X2 p(χ21 ≤ X 2 ) SNP645 4.51 1.17 14.86 0.00 SNP294 4.37 1.18 13.72 0.00 SNP9 -0.18 1.10 0.027 0.87 SNP194 6.07 1.24 23.96 0.00 SNP297 -0.20 1.09 0.034 0.86 fˆM ICEY Y Z se X2 p(χ21 ≤ X 2 ) 5.70 1.11 26.37 0.00 5.88 1.14 26.60 0.00 -0.49 1.04 0.22 0.63 4.92 1.63 9.11 0.00 0.17 1.04 0.027 0.87 fˆM ICEY Z Z se X2 p(χ21 ≤ X 2 ) 2.50 1.06 5.56 0.02 2.78 1.16 5.74 0.02 -0.38 1.08 0.12 0.72 4.44 1.57 7.99 0.00 -0.49 1.06 0.21 0.64 fˆCATY Y Z se X2 p(χ21 ≤ X 2 ) 5.96 1.07 31.03 0.00 5.91 1.27 21.66 0.00 0.51 1.13 0.20 0.65 4.45 1.15 14.97 0.00 -0.07 1.30 0.002 0.96 fˆM IXY Z Z se X2 p(χ21 ≤ X 2 ) 1.26 1.01 1.56 0.21 1.43 1.01 2.01 0.16 -1.11 1.06 1.09 0.29 1.20 1.49 0.65 0.42 -0.66 1.01 0.43 0.51 fˆM N AR1 Z se X2 p(χ21 ≤ X 2 ) 5.41 1.17 30.25 0.00 4.32 1.05 16.47 0.00 0.08 1.04 0.19 0.94 5.86 1.19 9.49 0.00 0.46 1.06 0.053 0.67 fˆM N AR2 Z se X2 p(χ21 ≤ X 2 ) 6.27 1.14 21.38 0.00 4.91 1.21 16.93 0.00 -0.49 1.11 0.006 0.66 6.01 1.95 24.25 0.00 0.25 1.09 0.19 0.81 Tabla 5.33: Combinación de estadı́sticos 5.8. NÚMERO DE MARCADORES SIGNIFICATIVOS BAJO IMPUTACIÓN 75 Podemos observar en la tabla 5.33 que el único modelo que no es significativo es el M IXY Z , es decir, aceptamos la hipótesis de que para estos SNPs analizados, ellos se encuentran bajo equilibrio. Todos los demás modelos rechazan equilibrio para los SNPs 645, 294 y 194 y aceptan equilibrio para 9 y 297. Esto evidencia que el hecho de aplicar un modelo determinado a un SNP este mismo no sea satisfactorio a el resto de los SNPs en cuestión de alcanzar HWE. Si nos remitimos a las figuras 5.10-5.14 ya habı́amos comentado que estos SNPs tiene sus categorı́as definidas de diferentes formas, esto puede indicarnos que quizás debemos ser más cuidadosos a la hora de escoger el método de imputación en dependencia de cómo están distribuidas las categorı́as en el “CallPlot” y tener en cuenta las medidas de las intensidades si queremos llegar a tener conteos bajo equilibrio. 5.8. Número de Marcadores significativos bajo imputación En la sección 5.1 expusimos el diagrama ternario de las composiciones genotı́picas de los SNPs observados completamente sobre la región de aceptación, donde hicimos referencia que de 376 SNPs se encontraron 23 de ellos que no cumplı́an con la condición de equilibrio de Hardy-Weinberg. Ahora realizaremos el plot ternario para todos los SNPs descartando los Missing. Figura 5.27: Diagrama ternario de las frecuencias genotı́picas para los SNPs descartando Missing. También habı́amos comentado que eliminamos 5 SNPs de los 1000 que estamos analizando por presentar más de un 50 % de casos no observados. Por lo tanto, la figura 5.27 76 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS representa el diagrama ternario de 995 SNPs donde 103 de ellos resultaron significativos, aproximadamente representa un 11 %. Si analizamos al nivel del 5 %, deberı́amos haber obtenido 50 SNPs significativos como máximo para poder decir que nuestra base de datos, descartando los missing por cada SNP, presentarı́a equilibrio de Hardy-Weinberg, donde la cifra obtenida es el doble de esta, lo cual indica que de manera global nuestros datos no están en equilibrio para este caso. También observamos que de nuevo se repite el patrón de déficit de heterocigotos. Para la imputación multivariada usamos el modelo M ICEY Z debido a que en el modelo M IXY Z hemos presentado problemas con el software y se está trabajando en base a ello. SNPs # de SNPs Sin Missing 376 # de Significativos Omitiendo Missing 23 % respecto al # de SNPs 6.11 % % de Significativos respecto al total 2.31 % Con Missing 619 80 12.92 % 8.04 % <10 % >10 % 522 97 51 29 9.77 % 29.9 % 5.12 % 2.92 % Total 995 103 58.70 % 10.35 % SNPs # de SNPs % respecto al # de SNPs 6.11 % % de Significativos respecto al total 2.31 % Sin Missing 376 # de Significativos Imputados 23 Con Missing 619 74 11.95 % 7.43 % <10 % >10 % 522 97 51 23 9.77 % 23.71 % 5.12 % 2.31 % Total 995 97 51.54 % 9.74 % Tabla 5.34: Comparativa de porcentajes respecto a omitir e imputar missing En la tabla 5.34 podemos observar que existe casi el doble de SNPs con Missing respecto a los SNPs observados completamente, dentro de aquellos no observados hemos estratificado 2 categorı́as, los SNPs con menos de un 10 % de Missing y aquellos SNPs con más de este mismo umbral. En la columna 4 observamos que el hecho de incrementar el # de missing aumenta el porcentaje de significativos respecto al número de SNPs, por lo tanto esto puede ser un indicador de evidencia de error en genotipado. Aunque esperábamos menos SNPs significativos, sin embargo, sabemos que imputando fˆ suele bajar, pero no siempre acaba de traspasar el umbral entre significativo y no-significativo en todos los SNPs, como bien veı́amos en la tabla 5.23 referente al método M ICEY Z . Según los resultados, probablemente con el modelo M IXY Z hubiéramos 5.8. NÚMERO DE MARCADORES SIGNIFICATIVOS BAJO IMPUTACIÓN 77 encontrado menos significativos. (a) 995 SNPs (b) < 10 Missing (c) > 10 Missing Figura 5.28: fcc vs fimp La figura 5.28 muestra lo descrito en la tabla 5.34. En el gráfico 5.28a tenemos la representación general de todos aquellos SNPs con Missing, en el gráfico 5.28b los SNPs con menos de 10 % de Missing y en el gráfico 5.28c aquellos con 10 % y más de Missing. Podemos observar que los marcadores con más 10 % Missing se encuentran en general por debajo de la recta y = x esto indica que la estimación del coeficiente de endogamia baja cuando se imputa. De manera general, es un indicador de que cuando se descartan los Missing las estimación de éste coeficiente está sesgado, es decir, se rechaza el equilibrio más a menudo de lo que se deberı́a. En los 3 gráficos, observamos los puntos de color verde y rojos; los verdes son aquellos para los cuales tanto el coeficiente de endogamia de los SNPs, eliminando los 78 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS casos no observados como imputados, pues no resultaron significativos y para los puntos rojos lo contrario. TopTen de los más significativos SNP110 SNP179 SNP197 SNP229 SNP240 SNP274 SNP280 SNP365 SNP370 SNP542 AA AB BB NA fcc fimp 5 1 75 5 41 78 52 80 0 1 36 10 3 28 34 14 41 13 15 31 56 79 0 66 13 3 6 1 84 67 2 9 21 0 11 4 0 5 0 0 0.80 1 0.82 1 1 0.93 0.81 0.79 1 1 0.80 1 0.82 1 1 0.88 0.81 0.79 1 1 En la tabla 5.35 consideramos los 10 SNPs más significativos con sus respectivas estimaciones del coeficiente de endogamia tanto descartando missing como imputándolos. Pudiéramos considerar que estos marcadores sean los candidatos más probables para declarar error de genotipado o bien que están más asociados a la enfermedad. Cabe indicar que estos SNPs más significativos no fueron los que más missing tenı́an. Tabla 5.35: Marcadores más significativos 5.9. Cálculo de la potencia y tamaño muestral A la luz de la base de datos estudiada en este capı́tulo, presentamos algunos cálculos respecto a la potencia de los tests. Hacemos la misma distinción entre las pruebas clásicas para HWE y pruebas para HWE en relación con una enfermedad genéticamente determinada como ya se ha presentado en la Sección 2.7. 5.9.1. Potencia de las pruebas clásicas de HWE Como mencionamos en la sección 2.7 podemos calcular la potencia del test χ2 dado un tamaño muestral, un nivel de significación y un grado de desequilibrio D, pero también a la inversa, es decir, el tamaño de la muestra necesaria para obtener una potencia dada. También podemos realizar el cálculo de la potencia para la prueba exacta dado el mı́nimo conteo alélico. En nuestra base de datos tenemos 99 individuos que bajo diferentes escenarios del grado de desequilibrio y conteos alélicos, podemos observar la potencia que adquieren el test Exacto y la prueba χ2 , a través de la figura 5.29. Estos cálculos revelan lo siguiente: 1. Bajo HWE, tenemos θ = 4. En esta situación, la potencia alcanza exactamente el error de tipo I. El gráfico 5.29c muestra que la clásica prueba de Chi-cuadrado puede exceder la tasa de rechazo nominal, en particular para las frecuencias de los alelos más bajas. 79 0.6 0.3 Power 0.0 0.0 0.1 0.2 0.2 0.4 Power 0.6 0.4 0.8 0.5 1.0 5.9. CÁLCULO DE LA POTENCIA Y TAMAÑO MUESTRAL 0 20 40 60 80 100 0 20 40 Minor allele count 60 80 100 80 100 Minor allele count (b) 0.00 0.02 0.04 Power 0.06 0.08 0.10 (a) 0 20 40 60 80 100 Minor allele count 1.0 0.8 0.6 Power 0.4 0.2 0.0 0.0 0.1 0.2 Power 0.3 0.4 0.5 (c) 0 20 40 60 80 100 0 20 Minor allele count 40 60 Minor allele count (d) (e) Figura 5.29: Test Exacto (rojo) y Test χ2 (verde) 80 CAPÍTULO 5. ANÁLISIS DE LOS RESULTADOS 2. La prueba exacta tiene una tasa de rechazo que siempre es inferior a la tasa nominal, y es a veces muy por debajo de la tasa nominal. Por lo tanto, la prueba exacta es una prueba conservadora de HWE. 3. La potencia suele ser mejor para una frecuencia alélica menor de 0.5, pero obtiene peores potencias para muy bajas frecuencias alélicas. 4. La prueba de chi-cuadrado tiene una potencia ligeramente mejor que la prueba exacta, pero la desventaja de esto es, como se ha mencionado antes, que incrementa la tasa de error de tipo I. 5. Para el tamaño de la muestra dada de n = 99, la potencia de detectar “moderadas ” desviaciones de HWE (θ = 2 o θ = 8) es baja, y en general no excederá el 0.4. 6. Para desviaciones extremas de HWE (θ = 1 o θ = 16), una potencia razonable de 0.8 se puede conseguir si la frecuencia del alelo es por encima de 0.25. 5.9.2. Potencia de la prueba de HWE para la asociación marcadoresenfermedad Figura 5.30: Potencia en función de la frecuencia alélica q 5.9. CÁLCULO DE LA POTENCIA Y TAMAÑO MUESTRAL 81 Cálculos para el tamaño de la muestra y una fórmula del cálculo de potencia para el test de HWE para asocación marcadores-enfermedad se han descrito por Lee [7] y se resumieron en la Sección 2.7. La figura 5.30 presenta las funciones de la potencia para el tamaño de la muestra de la base de datos estudiada en este capı́tulo, n = 99. Dos niveles de significación fueron utilizados, α = 0,05 (fila de paneles superiores) y α = 0,0001 (fila de paneles inferiores). Tomar α = 0,05 corresponde a un nivel de significancia estándar para probar un sólo marcador, mientras que 0.0001 es un nivel más estricto que se utiliza cuando muchos marcadores se ponen a prueba. El riesgo relativo dado en Lee [7] γ se fijó en 4, 2 y 1.5. La potencia fue calculada para los modos de herencia aditivo (azul), recesivo (verde) y dominante (rojo). La figura muestra que con alfa = 0.0001, la potencia de la prueba es muy baja, y no excede de 0.3, incluso con un fuerte efecto de enfermedad (γ = 4). La potencia es razonable (≥ 0,80), cuando α = 0,05 y un efecto de la enfermedad fuerte y frecuencias alélicas intermedias. Los modelos dominante y recesivo se consideran que tienen más potencia que el modelo aditivo. En el contexto del análisis de nuestra base de datos sobre el cáncer de colon, 1000 marcadores fueron probados, y una corrección para múltiples pruebas se indicó, a un nivel de significación de 0.0001 o incluso menor pudiera realizarse. Esto implica que, con los datos que tenemos, la potencia para detectar asociación marcadores-enfermedad por medio de una prueba de HWE es muy baja. Capı́tulo 6 Discusión y conclusión El objetivo de este trabajo ha sido realizar inferencia estadı́stica sobre el equilibrio de Hardy-Weinberg en presencia de datos genotı́picos faltantes. Para alcanzar este objetivo, la prueba clásica de chi-cuadrado para equilibrio se ha reformulado como un problema de estimación de parámetros, en este caso la estimación del coeficiente de endogamia. Nos planteamos evaluar la sensibilidad de este coeficiente a través de distintos procedimientos de sustitución de datos omitidos, es decir, inferencia sobre f para HWE teniendo en cuenta los datos faltantes. Indagamos sobre el tipo de patrón de los Missing Data, donde mostramos que estábamos en presencia de un patrón no-monótono, por lo que todos los procedimientos para la imputación de los datos faltantes se basaron en algoritmos de imputación múltiple. Debido a que este coeficiente es obtenido mediante el cálculo de las frecuencias alélicas y éstas por los conteos genotı́picos y que además tienen una relación estrecha con las medidas de las intensidades alélicas; nos postulamos varios modelos a imputar. En cada uno de ellos estudiamos el respectivo sesgo que producen las imputaciones respecto al análisis de los datos descartando los missings; el comportamiento de los métodos analizados respecto a los valores que tomaron y su tendencia a incrementar o decrementar nuestro parámetro de interés. No hay peor modelación que la que no se hace, por esto en nuestro estudio nos planteamos diferentes modelos bajo 2 categorı́as: Modelación Conjunta e Imputación Múltiple de Regresión Secuencial. La eficacia de los modelos utilizados en la imputación depende de las covariables incluidas. Se han incorporado las intensidades del marcador a imputar y otros marcadores 83 84 CAPÍTULO 6. DISCUSIÓN Y CONCLUSIÓN genéticos. Las intensidades resultaron ser predictoras fuertes de los marcadores a imputar. La inclusión de otros marcadores con datos completos como covariables cambiaba la estimaciones del coeficiente de endogamia. Los 5 marcadores escogidos no han sido los más adecuados para la imputación, por falta de conocimiento de su ubicación fı́sica. En trabajo futúro se considera utilizar solo marcadores fı́sicamente cercanos y correlacionados con el SNP a imputar ası́ como adicionar otras covariables de interés para la enfermedad del cáncer de colon, como la edad del paciente, antecedentes cancerı́genos entre otros. Para el estudio usamos diferentes paquetes implementados en el software R. Estos son MICE, CAT y MIX. A través del curso del estudio, hicimos comparaciones de las diferentes metodologı́as que usan cada paquete de estos. Llegamos a que la modelación usando MICE (imputación multivariada) y CAT (imputación univariada) incluyendo sólo SNPs tuvieron la misma tendencia, imputar sobre aquella categorı́a de mayor conteo. Los modelos implementados incluyendo las intensidades solamente a través de MICE y MIX, siguieron el mismo patrón de imputación, aumentar la categorı́a de los heterocigotos y los modelos donde incluimos tanto las intensidades como los SNPs observados y no observados, sus categorı́as se equilibraban. Como habı́amos comentado el principal objetivo de la imputación fue generar estimaciones del coeficiente de endogamia haciendo uso de criterios estadı́sticos, donde la elección del método debiera sustentarse en la sensibilidad de los estimadores. La teorı́a avala que los estimadores generados por los métodos de imputación múltiple son robustos y la sustitución de valores omitidos se realiza en forma estocástica, lo que garantiza que no se introducen sesgos de asignación. Las propiedades estadı́sticas de los estimadores se sustentan en técnicas bayesianas de probada utilidad, ası́ como en procedimientos estocásticos de cadenas de Markov. De las varias alternativas, vimos cuál de éstas completaba los datos faltantes y justificara mejor los fundamentos teóricos de los procedimientos aplicados, llegamos a que el Modelo de Localización General era la metodologı́a más eficiente implementado en el programa MIX. Sin embargo, la gran parte de las imputaciones se ha realizado utilizando el modelo multinomial logit implementado en el software MICE y queda pendiente resolver algunos problemas computacionales con el programa MIX. Vimos también que si la selección del método de imputación se sustenta únicamente en criterios estadı́sticos, como el análisis del error estándar, es posible concluir que cualquiera de las metodologı́as analizadas generan distribuciones equivalentes y que cualquiera podı́amos aplicar para la imputación general, pero a pesar de estas similitudes observamos que en dependencia del SNPs pues se debı́a aplicar una alternativa u otra. El análisis de sensibilidad realizado entre las diferentes metodologı́as de imputación 85 sobre MAR, obtuvimos que los resultados a través de Imputación Múltiple de Regresión Secuencial como con la Modelación Conjunta son muy similares. Ası́ como el análisis de sensibilidad entre mecanismos de respuestas MAR y MNAR, tuvieron estimadores muy cercanos. Coincidimos que tanto por la regla de Rubin para el pooling del estimador como a través del pooling de estadı́sticos se llega al mismo resultados, donde el modelo más eficiente es el aplicado a través del Modelo de Localización General. Se observo que, para SNPs con un porcentaje substancial de missings, el coeficiente de endogamia estimado mediante métodos con imputación múltiple fue en general, más bajo que la estimación obtenido descartando los datos faltantes. Eso surgiere que las pruebas para HWE que descartan missing pueden estar sesgadas. El número de SNPs significativos encontrado en el estudio es en general más alto de lo que se esperaba por efectos del azar solo. Es difı́cil valorar si esto es debido a la asocicación entre marcadores y enfermedad o a errores de genotipado. El hecho de haya más significativos entre los marcadores con muchos missings sugiere que los errores de genotipado es un factor importante. Aunque expusimos que el Modelo de Localización General fue en nuestro estudio el más efectivo, no debemos generalizar que este sea el mejor ya que existe evidencia reciente [29] que bajo la modelación no paramétrica las estimaciones pueden resultar más eficientes en estos tipos de datos. Por lo que sugerimos como un estudio futuro, la modelación bajo este esquema. Hemos usado del coeficiente de endogamia para el estudio, este hecho ha implicado que para la inferencia sobre equilibrio usáramos la prueba clásica de chi-cuadrado. En la actualidad, las pruebas exactas se han puesto de moda. Otra vı́a de hacer inferencia para HWE en presencia de missings, que ha quedado pendiente de explorar, es mediante la combinación de pruebas exactas de juegos de datos imputados. Sugerimos que para estudios futuros, principalmente estudios de casos-controles, se realice el análisis de sensibilidad basados en los riesgos relativos genotı́picos para el caso de esta enfermedad. Existe evidencia que estos riesgos tienen alta relación con el desequilibrio de Hardy-Weinberg y este desequilibrio con el tamaño muestral; por lo que se debe escudriñar en este perfil para una mejor conclusión referente al equilibrio de Hardy-Weinberg [7]. Una vez imputados los missings, surgió la pregunta de cuál era la potencia de los tests utiltizados, tanto para las pruebas clásicas para equilibrio como para una prueba 86 CAPÍTULO 6. DISCUSIÓN Y CONCLUSIÓN HWE orientado a detectar asociación con la enfermedad. Se ha cuantificado en ambos casos la potencia de las pruebas HWE para una muestra como la observada. A la vista del gran número de marcadores en estudio, se considera que la potencia de los test para HWE es baja en ambos casos y que se necesitarı́an tamaños muestrales más grandes. Bibliografı́a [1] Wikipedia, “Single nucleotide polymorphic,” journal Wikipedia, vol. 1, p. 1, 2012. [cited at p. -] [2] [3] B. S. Weir, Genetic Data Analysis II, Massachusetts, Ed. Sinauer Associates, Inc, 1996. [cited at p. -] Wikipedia, “Hardy weinberg equilibrium,” journal Wikipedia, vol. 1, p. 1, 2012. [cited at p. -] [4] T. Emigh, “Comparison of tests for hardy-weinberg equilibrium,” journal Biometric, vol. 36, p. 627642, 1980. [cited at p. -] [5] R. Rohlfs and B. Weir, “Distributions of hardy-weinberg equilibrium tests statistics,” journal Genetics Society of America, vol. 180, pp. 1609–1616, September 10, 2008. [cited at p. -] [6] J. Graffelman, “The hardy-weinberg package.” software. [7] W. C. Lee, “Searching for disease-susceptibility loci by testing for hardy-weinberg disequilibrium in a gene bank of affected individuals,” American Journal of Epidemiology, vol. 158; 5, pp. 1–5, 2003. [cited at p. -] [8] N. H. G. R. Institute, “A catalog of published genome-wide association studies,” Genome.Gov, vol. 1, p. 1, 2012. [Online]. Available: www.genome.gov/ gwastudies/index.cfm?pageid=26525384/searchForm [cited at p. -] [9] C. S. Piè, “Study and validation of data structures with missing values. application to survival analysis.” Ph.D. dissertation, Universitat Politècnica de Catalunya, 2001. [cited at p. -] [cited at p. -] [10] D. Rubin, Multiple imputation for nonresponse in surveys, V. Barnett, R. A. Bradley, J. S. Hunter, and D. G. Kendall, Eds. John Wiley & Sons, Inc., 1987. [cited at p. -] 87 88 BIBLIOGRAFÍA [11] R. J. Little and D. B. Rubin, Statistical Analysis with Missing Data, V. Barnett, R. A. Bradley, J. S. Hunter, and D. G. Kendall, Eds. John Wiley & Sons, Inc., 1987. [cited at p. -] [12] P. Zhang, “Multiple imputation: Theory and method,” International Statistical Review, vol. 71, pp. 581–592, 2003. [cited at p. -] [13] Y. Y. SHIEH, “Imputation methods on general linear mixed models of longitudinal studies,” journal Biometric, vol. 1, p. 1, 2000. [cited at p. -] [14] F. Medina and M. Galván, “Imputación de datos: Teorı́a y práctica,” CEPAL, vol. 54, pp. 1–84, 2007. [cited at p. -] [15] J. L. Schafer, Analysis of incomplete multivariate data., Chapman and Hall, Eds. Chapman and Hall., 1997. [cited at p. -] [16] M. G. K. Geert Molenberghs, Missing Data in Clinical Studies, S. S. Vic Barnett, Ed. John Wiley & Sons, Ltd., 2007. [cited at p. -] [17] V. B. S, “Multiple imputation of discrete and continuos data by fully conditional specification,” Statistics Methods in Medical Research, vol. 16, pp. 219–242, 2007. [cited at p. -] [18] Y. He, “Missing data analysis using multiple imputation getting to the heart of the matter,” Ph.D. dissertation, Harvard Medical School, 2010. [cited at p. -] [19] S. V. Buuren and K. Groothuis-Oudshoorn, “Package mice: Multivariate imputation by chained equations in r,” Journal of Statistical Software, vol. VV, pp. 1–68, 2012. [Online]. Available: www.stefvanbuuren.nl [cited at p. -] [20] M. A. Tanner and W. H. Wong, “The calculation of posterior distributions by data augmentation,” Journal of the American Statistical Association, vol. 82, No 398, pp. 528–540, 1987. [cited at p. -] [21] D. B. Rubin, “The calculation of posterior distributions by data augmentation. comment: A noniterative sampling/importance resampling. alternative to the data augmentation algorithm for creating a few imputations when fractions of missing information are modest: The sir algorithm.” Journal of American Statistical Association., vol. 82, pp. 543–546, 1987. [cited at p. -] [22] J. Graffelman and J. M. Camarena, “Graphical tests for hardy-weinberg equilibrium based on the ternary plot,” Human Heredity, vol. 65, p. 7784, 2008. [cited at p. -] [23] S. V. Buuren, H. Boshuizen, and D. Knook, “Multiple imputation of missing blood pressure covariates in survival analysis,” Statistics in Medicine, vol. 18, pp. 681– 694, 1999. [cited at p. -] BIBLIOGRAFÍA 89 [24] U. A. T. Services, “What is complete or quasi-complete separation in logistic/probit regression and how do we deal with them?” Software product by the University of California., vol. 1, p. 1, 2012. [Online]. Available: www.ats.ucla. edu/stat/mult pkg/faq/general/complete separation logit models.htm [cited at p. -] [25] J. L. Schafer, “Multiple imputation: A primer.” Statistics Methods in Medical Research, vol. 8, pp. 3–15, 1999. [cited at p. -] [26] ——, “Package cat: Analysis of categorical-variable datasets with missing values,” Statistics Methods Software R, vol. 1, p. 1, 2012. [Online]. Available: www.stat.psu.edu/∼jls/misoftwa.html/aut [cited at p. -] [27] ——, “Package mix: Estimation/multiple imputation for mixed categorical and continuous data,” Statistics Methods Software R, vol. 1, pp. 1–15, 2012. [Online]. Available: www.stat.psu.edu/∼jls/misoftwa.html [cited at p. -] [28] S. R. Cook, “Using historical data to model and impute long-term disease progression,” Ph.D. dissertation, Department of Statistics. Columbia University, 2004. [Online]. Available: www.stat.columbia.edu/∼cook/penn.pdf [cited at p. -] [29] D. J. Stekhoven and P. Bühlmann, “Missforest - nonparametric missing value imputation for mixed-type data,” Oxford Journal’s Bioinformatics, vol. This article has been submitted, pp. 1–13, 2011. [Online]. Available: www.stat.ethz.ch/CRAN [cited at p. -] Índice de figuras 1.1. Comparación entre ADN’s . . . . . . . . . . . . . . . . . . . . . . . . . . 2.1. 2.2. 2.3. 2.4. 2.5. 2 SNPs mostrando variabilidad entre individuos de una misma especie Un individuo que es heterocigoto para un determinado SNP . . . . . . Hardy (arriba) Weinberg (abajo) . . . . . . . . . . . . . . . . . . . . frecuencias de Hardy-Weinberg . . . . . . . . . . . . . . . . . . . . . Distribución de los heterocigotos . . . . . . . . . . . . . . . . . . . . . . . . . 8 8 9 10 16 3.1. Cáncer de Colon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.2. Estadı́os del cáncer de Colon . . . . . . . . . . . . . . . . . . . . . . . . . 23 24 4.1. 4.2. 4.3. 4.4. 4.5. (a) Medida de Intensidad A, (b) Medida de Intensidad B, (c) Genotipado Un patrón monótono de Missing Data . . . . . . . . . . . . . . . . . . Un patrón no monótono de Missing Data . . . . . . . . . . . . . . . . . Imputación Múltiple . . . . . . . . . . . . . . . . . . . . . . . . . . . Conjunto Datos con Missing Data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 31 31 33 36 5.1. 5.2. 5.3. 5.4. 5.5. 5.6. 5.7. 5.8. 5.9. 5.10. 5.11. 5.12. 5.13. MAF . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Q-Q Plots de los 3 test . . . . . . . . . . . . . . . . . . . . . Diagrama ternari y ScatterPlots de las frecuencias genotı́picas Conteo de Missing por SNPs . . . . . . . . . . . . . . . . . . Conteo de Missing por Individuos . . . . . . . . . . . . . . . Diagramas bivariantes de intensidades para 4 SNPs . . . . . . Pruebas Mecanismo de los Patrones Missing Data . . . . . . . Proporción de Missing y Combinaciones . . . . . . . . . . . . Plots Marginales . . . . . . . . . . . . . . . . . . . . . . . . SNP 645 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . SNP 294 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . SNP 9 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . SNP 194 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41 42 44 45 45 46 48 50 51 52 52 52 52 91 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 92 ÍNDICE DE FIGURAS 5.14. SNP 297 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 5.15. Evidencia de sesgo entre la Imputación Simple y los Casos Observados . . 53 5.16. Regresión Logı́stica Multinomial . . . . . . . . . . . . . . . . . . . . . . . 55 5.17. Método Regresión Lineal Bayesiana . . . . . . . . . . . . . . . . . . . . . 57 5.18. Modelo M ICEY Y Z . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58 5.19. Modelo M ICEY Y . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58 5.20. Modelo M ICEY Z . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59 5.21. Evidencia de sesgo en las imputaciones realizadas al SNP645 . . . . . . . . 60 5.22. Evidencia de sesgo en las imputaciones para diferentes SNPs . . . . . . . . 61 5.23. Evidencia de sesgo en las imputaciones para diferentes SNPs . . . . . . . . 63 5.24. Evidencia de sesgo en las imputaciones para diferentes SNPs . . . . . . . . 65 5.25. Posiciones relativas para los estimados de fˆ. cc: Casos Observados. is: Imputación Simple. m1: M ICEY Y Z . m2: M ICEY Y . m3: M ICEY Z . m4: CATY Y . m5: M IXY Z . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 5.26. Posiciones relativas para los estimados de fˆ. cc: Casos Observados. is: Imputación Simple. m1: M ICEY Y Z . m2: M ICEY Y . m3: M ICEY Z . m4: CATY Y . m5: M IXY Z . m6: M N AR1 . m7: M N AR2 . . . . . . . . . . . . . . . . 73 5.27. Diagrama ternario de las frecuencias genotı́picas para los SNPs descartando Missing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75 5.28. fcc vs fimp . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77 5.29. Test Exacto (rojo) y Test χ2 (verde) . . . . . . . . . . . . . . . . . . . . . 79 5.30. Potencia en función de la frecuencia alélica q . . . . . . . . . . . . . . . . 80 Índice de tablas 2.1. En esta tabla vemos que los tamaños de muestra son necesarios a fin de obtener una potencia del 80 % para la detección de asociación mediante la prueba de HWE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 4.1. Eficiencia relativa ( %) de la estimación mediante Imputación Múltiple por número de imputaciones y fracción de información Missing . . . . . . . . . 35 5.1. SNPs que menos Observaciones aportaron 5.2. SNPs que más Missing aportaron . . . . . 5.3. Regresión Logı́stica Multinomial . . . . . 5.4. Matriz Predictor . . . . . . . . . . . . . . 5.5. MultiLogit M ICEY Y Z . . . . . . . . . . 5.6. MultiLogit M ICEY Y . . . . . . . . . . 5.7. MultiLogit M ICEY Z . . . . . . . . . . 5.8. M ICEY Y Z . . . . . . . . . . . . . . . . 5.9. M ICEY Y . . . . . . . . . . . . . . . . . 5.10. M ICEY Z . . . . . . . . . . . . . . . . . 5.11. CATY Y . . . . . . . . . . . . . . . . . . 5.12. M IXY Z . . . . . . . . . . . . . . . . . . 5.13. M IXY Z . . . . . . . . . . . . . . . . . . 5.14. M ICEY Z . . . . . . . . . . . . . . . . . 5.15. Descriptiva . . . . . . . . . . . . . . . . 5.16. M ICEY Y Z . . . . . . . . . . . . . . . . 5.17. M ICEY Y . . . . . . . . . . . . . . . . . 5.18. M ICEY Z . . . . . . . . . . . . . . . . . 5.19. CATY Y . . . . . . . . . . . . . . . . . . 5.20. M IXY Z . . . . . . . . . . . . . . . . . . 5.21. Modelo M ICEY Y Z . . . . . . . . . . . 5.22. Modelo M ICEY Y . . . . . . . . . . . . 5.23. Modelo M ICEY Z . . . . . . . . . . . . 49 49 55 56 58 58 59 60 60 60 63 64 65 65 65 66 66 66 66 66 67 67 67 93 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94 ÍNDICE DE TABLAS 5.24. Modelo CATY Y . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.25. Modelo M IXY Z . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.26. Resumen de las variables que son usadas para la imputación. Las columnas de la 2-6 contiene las correlaciones de las variables filas respecto a los SNPs Missing. Columna 7 es un ejemplo de la correlación entre el indicador de respuesta y los datos del SNP 645. Columna 8 es el porcentaje de casos usables que es igual al porcentaje de los datos observados de las variables filas entre el subgrupo de casos que tienen Missing para el SNP 645 . . . . 5.27. Matriz Predictora: MNAR . . . . . . . . . . . . . . . . . . . . . . . . . . 5.28. M N AR1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.29. M N AR1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.30. M N AR2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.31. M N AR2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.32. Errores Estándares . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.33. Combinación de estadı́sticos . . . . . . . . . . . . . . . . . . . . . . . . . 5.34. Comparativa de porcentajes respecto a omitir e imputar missing . . . . . . 5.35. Marcadores más significativos . . . . . . . . . . . . . . . . . . . . . . . . 67 68 71 72 72 72 72 72 73 74 76 78