Clasificación y manifestaciones clínicas de las enfermedades
Anuncio

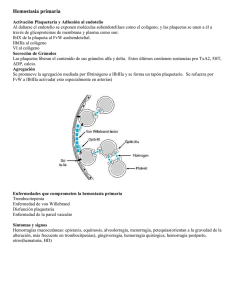
Clasificación y manifestaciones clínicas de las enfermedades hemorrágicas Dr. Oswaldo Carrasquel Servicio de Banco de sangre y Hematología Hospital Dr. Raúl Leoni Importancia de la historia clínica 1. ¿Existe una tendencia hemorrágica? 2. ¿Es familiar o adquirida la alteración? 3. ¿Es la tendencia hemorrágica inducida por algún fármaco? 4. ¿Existe alguna otra enfermedad o situación que puede ser la causa o puede exacerbar la tendencia hemorrágica? 5. ¿Afecta la alteración a la hemostasia primaria o a la formación y estabilización de la fibrina? Clasificación Hereditarios Adquiridos Mecanismo: Vascular Plaquetario Coagulación Fibrinolisis Combinación Trastornos hereditarios Deficiencias de factores de la coagulación Defectos plaquetarios Defectos en la fibrinolisis Vascular: Telangectasias hemorrágicas Enf. tejido conectivo: Sindrome de Ehlers Danlos Trastornos adquiridos Trombocitopenias Enfermedad hepática Insuficiencia renal Enfermedades hematológicas Deficiencia de vitamina K Inhibidores adquiridos CID Medicamentos Vascular Aspectos Clínicos Localización de la hemorragia Edad de aparición Historia familiar: patrón de herencia o consanguinidad Forma de presentación Condiciones del paciente Enfermedades asociadas Clínica Sangrado excesivo en exodoncias, cirugías Equímosis o hematomas no acordes a trauma Hemartrosis Epistaxis, > 15 min, recurrente que no cesa con presión Menstruación mayor de 7 días Sangrado que haya requerido transfusión Medicamentos Trastornos hemorrágicos CARACTERÍSTICAS DIFERENCIALES I Clínica Defecto de coagulación Defecto plaquetario Sangrado por heridas superficiales No excesivo Profuso y prolongado Sangrado por heridas profundas Comienzo retardado, contínuo, no cede con medidas locales Comienzo inmediato, cede con medidas locales Trastornos hemorrágicos CARACTERÍSTICAS DIFERENCIALES II Clínica Defecto de coagulación Defecto plaquetario Equimosis y hematomas Profundos y aislados Pequeños, superficiales, múltiples Hemartrosis Frecuentes Raras Trastornos hemorrágicos CARACTERÍSTICAS DIFERENCIALES III Clínica Defecto de coagulación Defecto plaquetario Síntomas más característicos Hemorragias tisulares (articulaciones, músculos) sangrado excesivo después del trauma Epistaxis, menorragia, púrpura, equímosis Clasificación Mucocutáneo Inmediato Defectos de hemostasia primaria No mucocutáneo Tardio Defectos de la coagulación o de la fibrinolisis Localización del sangrado Gingivorragia Epistaxis Menorragia Petequias Desórdenes plaquetarios Enf. von Willebrand Hemartrosis Sangrado muscular Cefalohematomas, HIC Cordón umbilical Déficit de factores de la coagulación SINTOMAS COMUNES DE LA HEMOFILIA Pueden presentarse hemorragias en cualquier parte del cuerpo; a veces visibles y a veces no. La hemorragia puede ocurrir después de una lesión o una cirugía. También ocurre sin motivo aparente es la llamada hemorragia espontánea. Los sangrados son infrecuentes en bebés con hemofilia, aunque puede aparecer luego de realizar un procedimiento invasivo. Cuando los niños empiezan a caminar, se golpean con facilidad. También sangran más tiempo de lo normal posterior a una lesión, particularmente en la boca y la lengua. Con el crecimiento, las hemorragias espontáneas son más comunes; afectando las articulaciones y los músculos. Hemostasia: Trastornos adquiridos Trombocitopenias Enfermedad hepática Insuficiencia renal Enfermedades hematológicas Deficiencia de vitamin K Inhibidores adquiridos CID Medicamentos Vascular Clasificación de los desórdenes plaquetarios Trombocitopenia Trombocitopatías o disfunción plaquetaria Hereditarias o Adquiridas Desòrdenes plaquetarios :Hereditarios • Número de plaquetas: MYH9, Tp Amegacariocítica congénita y variantes • Función específica: S. Bernard Soulier, Trombastenia Glanzmann, SWA • Gránulos plaquetarios Granulos densos, HPS, CHS, plaqueta gris,S Quebec • Exposición de fosfolìpidos: S Scott • Receptores y señales intraplaquetarias: DCOX, TxS, TxA2R, Receptor de ADP (PY12) Disfunción plaquetaria adquirida Desórdenes mieloproliferativos Trombocitemia esencial Policitemia vera LMC Uremia Hepatopatías CID Metaplasia mieloide Leucemias agudas y Mielodisplasia Defecto de secreción: Asociado a Eosinofilia o Anticuerpos antiplaquetarios Cirugía cardiovascular Agentes farmacológicos Manifestaciones Clínicas de los trastornos plaquetarios Púrpura: Puede ser signo temprano, período neonatal Epistaxis Gingivorragia Sangrado menstrual profuso o post-partum Hemorragia gastrointestinal, Hematuria Excepcionales: Hematomas viscerales o intramusculares Defectos de Adhesión: Síndrome de Bernard Soulier Herencia: Autosómica recesiva Características: - TS prolongado Trombocitopenia Plaquetas gigantes Agregación con ristocetina y Fib bovino ME: vacuolas y complejos de membrana Base molecular: Def. cuantitativa o cualitativa de GP Ib-IX Defecto de agregación plaquetaria: Trombastenia de Glanzmann Herencia: Autosómica recesiva Características: - - TS Agregación con Adrenalina, ADP y Colágeno Retracción del coágulo variable Base molecular: - Complejo GP IIb-IIIa - Deficiencias cuantitativas total (I) o parcial (II) - Alteración cualitativa (Variante) Defectos de la secreción plaquetaria Enfermedad del “pool” de almacenamiento Herencia: Autosómica recesiva Características: • • TS variable Agregación: o o o o o Ausencia de 2da fase con adrenalina y ADP Colágeno Variable con Acido Araquidónico Reacción de liberación de gránulos anormal Ausencia de gránulos densos Asociación con S.de Hermansky-Pudlack, Chediak-Higashi, TAR y S. Agentes farmacológicos que afectan la función plaquetaria Mecanismo Síntesis de TXA2 Receptor de ADP Receptor GPIIb-IIIa Aumento de AMPc o GMPc Variable Agente ASA, AINEs Ticlopidina, Clopidogrel Abciximab, Tirofiban, etc PI2, Dipiridamol, Xantinas, Cafeína, Teofilina, etc. Antimicrobianos Drogas de acción cardiovascular Anestésicos, Quimioterapia Antihistamínicos,Vitamina E Alimentos Enfermedad de von Willebrand Es un trastorno hemorrágico causado por defectos hereditarios en la concentración, estructura o función del FvW, independiente de cual gen o genes estén involucrados. Caracterizada por gran heterogeneidad genética, clínica y de laboratorio Sadler EJ. ISTH 2005. Szántó T et al. Sem Thromb Hemostasis 2012. EvW: Epidemiología Estudios de despistaje en población Italia: 0, 83% USA: 1,3% Francia: 0,6% Tipo 1 vs Posible Tipo 1 Prevalencia general: 74 (0,2-458) por millón Sintomáticos: 35 – 110 por millón Tipo 3: 0,5 - 6 por millón Rodeghiero F. 1987; Werner EJ; Biron C 1999; Favaloro E. 2011. Sadler E 2000 Clasificación de la EvW Tipo 1 Tipo 2 Tipo 3 Deficiencia cuantitativa parcial del FVW. Incluiría el Tipo 1 C Deficiencia cualitativa del FVW Deficiencia virtualmente completa del FVW Sadler J . 1994, 2006 Variantes del tipo 2 de la EvW 2A Disminución de las funciones dependientes de plaquetas asociado con ausencia selectiva de los multímeros de alto e intermedio PM 2B Aumento de la afinidad por la GPIb 2M Disminución de las funciones dependientes de plaquetas con multímeros de alto PM normales 2N Disminución marcada de la afinidad por FVIII Prevalencia de los tipos de EvW Tipo 1 60 – 80% Tipo 2 7 – 30% Tipo 3 5 – 20% Genética en EVW Tipo 1 Herencia dominante Oligogénica: Gene FVW, locus ABO y otros. Penetrancia y expresión variable Tipo 2 A, 2B, y 2M Herencia dominante, penetrancia completa Monogénica: gene FVW, penetrancia total Fenotipo similar : Pseudo VW o plaquetario por defecto en gen GPIBA Tipo 2 N y tipo 3 Herencia recesiva Monogénica: gene FVW Manifestaciones clínicas Jóvenes Edad avanzada Cavidad oral Gastrointestinal:angiodisplasias Epistaxis, equímosis Cirugía Sangrado menstrual, postparto Post exodoncia Heridas menores Hemartrosis Postexodoncia o cirugía Hematuria Niños: hemorragias conjuntivales, post-venipuntura, cefalohematoma Sangrado aumenta con la edad, especialmente en mujeres con EVW tipo 2 o 3 De Wee, E. Thromb Haemostas 2012. Miesbach W, Berntorp E. Eur J Haematol 2011. Bowman M. J Thromb Haemosta 2009 Diagnóstico de laboratorio Antígeno VWF:AG: ELISA (0-5U/dL), LIA (5-10 U/dL) Adhesión Bleeding time, PFA-100 MAPM función Cofcator Ristocetina y Unión a Colageno FVIII unión VWF:FVIIIB ELISA Panel básico VWF:Ag (ELISA), VWF:RCo, VWF:CB, FVIII y sus cocientes Pruebas adiconales PT, APTT, hematología, PFA-100, TS Otras APIR VWF Multimeros VWF:FVIIIB Caracterización de fenotipo Tipo de VWD PERFIL 1 Bajo VWF, con concordancia funcional. Razón WVF f/Ag ~ 1. > VWF 20U/dL APIR normal 2A Pérdida de MAPM. Bajo VWF con discordancia funcional Razón R Co/Ag, CB/Ag <0.7 2B Bajo a normal nivel VWFv usualmente con discordancia funcional Razón R Co/Ag, CB/Ag <0.7. Pérdida de MAPM y Trombocitopenia 2M Bajo a normal nivel VWF usualmente con discordancia funcional Razón R Co/Ag <0.7 and CB/Ag normal. MAPM presentes 2N Definido por prueba VWF:FVIIIB. Razón FVIII/VWF < 0.7 3 Tipicamente definido por VWF niveles <1% y FVIII < 10% Diagnostico de laboratorio EvW 3 y formas severas de EvW 1: FVW:RCo < 10 U dL + FVWAg- FVIII < 20 U dL Leves: F VWAg, FVIII: 20-50 U dL FVW:RCo 10-30 U dL Leves o no sangradores ? F VW:RCo, FVW:Ag, F VIII: 30-60 UdL Diagnostico de laboratorio Discrepancia entre resultados de variables estudiadas Tipo 2: FvW:RCo o FvW: CBA / FvWag < 0,7 Agregación con Ristocetina: 0,2 - 0, 6 mg/ml Tipo 2B Trombocitopenia Tipo 2B F VIII C / F vWag <0,7 Tipo 2N Tratamiento Factores a considerar Tipo de EVW; Nivel de FVIII y FVW. Respuesta previa a DDAVP Historia previa de sangrado Episodio a tratar Presencia de inhibidor Riesgos potenciales: acumulación de FVIII Tratamiento de la EvW Desmopresina Concentrados de F VIII/FVW o de FVW Crioprecipitados? Concentrados plaquetarios? Antifibrinolíticos Anovulatorios DDAVP. Indicaciones en EvW Establecida Tipo 1 Posible Tipo 2A, 2M, 2N Dudosa Tipo 2B, 3 Tratamiento con DDAVP Uso Contraindicaciones Intravenosa :0.3 µg kg-1, en 30 –50 ml salina infusión en 30 minutos Intranasal: adulto 300µg, niños 150 Menores de 2 años Mayores de 70 años Enfermedad arteriovascular Sindrome convulsivo Dosis se puede repetir en 12–24h Análisis de la respuesta individual Concentrados de FVIII + F vW Haemate P: Pasteurizado FvW:FVIII ≈ 2 Alphanate- FANHDI: SD + calor FvW:FVIII ≈ 1- Otros: Koate DI, Inmunate- Octanate Wilfactin, Wilate Tratamiento con concentrados Dosis IU/kg Frecuencia horas FVIII o FVWRCo IU/dL Cirugía mayor Cesarea 50 24-48 80-100 (2d) >50 (5-7d) Cirugía menor 30 24-48 > 30 (5-7d) Parto/Puerperio Anestesia epidural 50 Una dosis Exodoncias 20-40 1 Dosis > 30 -50 x 12 h Espontáneo 20-60 1 / día 2 -4 d > 30-50 Profilaxis 20-40 Problema 2 -3 /semana > 50 . (3-4d) >50 Según clínica Tratamiento: Complicaciones DDAVP: Taquifilaxia, enrojecimiento, hiponatremia, convulsiones, trombosis Concentrados F VIII+FvW : TEV por incremento del F VIII. EVW Tipo 3: Desarrollo de aloanticuerpos Makris M 2002; Mannucci P 2002 EvW en la mujer Menorragia y Hemoperitoneo por ovulación (EvW 3) Embarazo: Evaluación inicial y cerca de las 36 semanas FVIII < 50 U dL requiere tratamiento Precaución en puerperio por descenso de FVW-FVIII

![EL QUERUBICÓN VWfH ]ggf fH]g Lf f( ]g f g g]f $ f^h]h](http://s2.studylib.es/store/data/006120854_1-3c961fcdcedd4c447903f3665d2b3fc5-300x300.png)








