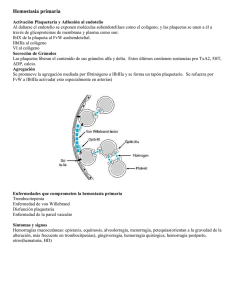
Rev Hematol Mex 2006 Vol 7 No 1 - Agrupación Mexicana para el
Anuncio

1 Control de Calidad en el Laboratorio de Hemostasia. María de los Angeles Ochoa-Rico Banco Central de Sangre, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, México, D.F., México. INTRODUCCIÓN. Los primeros avances sobre la explicación de la coagulación sanguínea se iniciaron a mediados de 1800, encontrando una relación de la coagulación sanguínea a problemas hemorrágicos, de flebitis, trombosis arterial y embolismo. El sistema hemostático se conforma por el endotelio vascular, plaqueta, factores de la coagulación y el sistema fibrinoliticos (1). Los procedimientos de control de calidad en el laboratorio son importantes en su organización y funcionamiento. Su objetivo es proporcionar exámenes confiables, reproducibles, exactos y ser por sí mismos relevantes para el diagnóstico y vigilancia clínica de los pacientes. Para lograr estos objetivos se requiere de una administración experta que supervise el trabajo del laboratorio, que garantice que se logre el nivel necesario de las buenas prácticas de laboratorio y que éste se mantenga constantemente, para lo cual es necesario llevar a cabo un programa de aseguramiento de calidad. Este programa contempla 3 aspectos (2): 1. Fase preanalítica. 2. Fase analítica. 3. Fase postanalítica. FASE PREANALÍTICA. Es la etapa del estudio que incluye: a) Solicitud de laboratorio. b) Indicaciones al paciente. c) Identificación del paciente. d) Sitio de punción. e) Manejo de la muestra. f) Transporte y almacenamiento. a) Solicitud de Laboratorio. Las muestras deben ir acompañadas de una solicitud debidamente formulada con la siguiente información: nombre completo, número de registro, edad, sexo, origen étnico, diagnóstico, medicamentos que recibe, última dosis de medicamentos anticoagulantes, inhibidores de fibrinolisis o fibrinolíticos (cuadro 1) (2). b) Indicaciones al paciente. El paciente debe presentarse a la toma de muestra con un ayuno de cuando menos 4 horas. La última ingesta de alimentos debe de ser baja en grasas, ya que la lipemia produce turbidez que interfiere con los métodos coagulómetricos y nefelométricos (3). c ) Identificación correcta del paciente. Es de primordial importancia etiquetar cada muestra en presencia del paciente con información suficiente para evitar confusión con otras muestras. Obtener una muestra de sangre de un paciente es mucho más que insertar una aguja en la vena o extraer una gota de sangre de un Solicitud de reimpresos: Dra. María de los A. Ochoa-Rico, Banco Central de Sangre, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, Av. Cuauhtémoc No. 330, Col. Doctores, C.P. 06720, México, D.F., México. Tel. 55 19 20 63 Revista de Hematología Vol. 7, No. 1, 2006 2 M de los A Ochoa-Rico. Cuadro 1 Fármacos que afectan las pruebas de laboratorio. Prueba Medicamento Efecto Factores II, V, VII, VIII IX, X, XII Anticonceptivos orales Aumentan la actividad Adhesión y agregación Plaquetaria Anticonceptivos orales Aumentan la actividad Tiempo de protrombina Anticoagulantes orales Cumarinicos Interfieren con las enzimas que reducen la vitamina K y limitan el proceso de caboxilación (TP alargado) TTPa o TT Heparina no fraccionada Inhibe al Factor X y II (TTPa alargado) Plaquetas Aspirina y antiinflamatorios no esteroides Antiagregantes.Inhiben a la ciclooxigenasa para la síntesis de tromboxano A2. dedo. Es el primer eslabón de una cadena de eventos que se completan cuando el medico recibe los resultados de las pruebas de su paciente y es el principio del control de calidad en el laboratorio clínico (4). La muestra debe tomarse correctamente y bajo condiciones favorables. El paciente debe estar tranquilo, relajado, ya que el estrés y el ejercicio afecta los factores de coagulación, así como la liberación de plaquetas a la circulación Cuadro 2 Alteraciones causadas por el estrés y ejercicio. Prueba Efecto Factor VIII Aumenta su nivel en plasma Función plaquetaria Afecta la 2a. fase de la agregación por ADP y Epinefrina Cuenta de plaquetas Aumenta Fibrinolisis Aumenta Revista de Hematología Vol. 7, No. 1, 2006 (cuadro 2) d) Seleccionar el sitio de punción. Elegir una vena de fácil acceso, como es la vena central, cefálica o radial del antebrazo, evitar áreas de hematomas o de cicatrización extensa. Se le pide al paciente que cierre el puño y se le frota el brazo de abajo hacia arriba, con objeto de hacer más visibles las venas. Debe evitarse el ejercicio excesivo de la mano porque aumentan los factores de coagulación. La punción debe ser limpia y única y sin exceder del tiempo de ligadura de un minuto; quitar la ligadura tan pronto como la sangre comience a fluir para evitar la estasis local (cuadro 3). e) Recolección de la muestra. El anticoagulante de elección es el citrato de sódio al 3.8% (0.129 M), en una proporción 9 partes de sangre y 1 de anticoagulante, recomendada por el Comité de Trombosis y Hemostasia (5). En tubos de plástico o de vidrio siliconizado, una vez obtenida la muestra, debe 3 Control de calidad en hemostasia. Cuadro 3 Alteraciones causadas por estasis venosa prolongada. Causa Efecto Liberación de Factor Tisular Activación de proteínas procoagulantes y anticoagulantes Hemólisis Liberación de fosfolipido activación de factores Salida de Plaquetas Activación ser mezclada con el anticoagulante mediante movimientos de inversión. Se debe evitar la hemólisis de la muestra, debido a que los eritrocitos liberan factores tromboplásticos al medio, que afectan acortando los tiempos de coagulación. Si el paciente tiene hematocrito menor de 30%, no es necesario hacer la corrección de la relación sangre anticoagulante, puesto que el sistema tiene suficiente calcio para evitar la coagulación. Pero cuando el hematocrito es mayor de 60%, los resultados son afectados por el exceso de anticoagulante, ya que puede quelar el calcio que se emplea durante el procedimiento de las pruebas de coagulación, causando prolongación de los tiempos de coagulación (6). Por ello, el volumen de anticoagulante debe ser ajustado para tener en cuenta la disminución del volumen plasmático siguiendo las pautas del cuadro 4, para obtener el volumen necesario de anticoagulante para una muestra de sangre de 5 mL. Otra forma de efectuar la corrección en pacientes que tengan valores de hematocrito mayor de 60% es empleando la siguiente formula: mL anticoagulantes(100 – Hto del paciente ) Volumen plasmático normal = mL de anticoagulante para mL de sangre Cuadro 4 Relación sangre anticoagulante con respecto al hematocrito. Hematocrito Volumen de Volumen de % anticoagulante sangre mL mL 60 70 80 0.4 0.25 0.2 4.5 4.75 4.8 Ejemplo: 0.3 x (100 -67) =0.18 mL anticoagulante + 2.7 mL sangre e) Manejo de la muestra. La estabilidad de las pruebas de coagulación es crítica para el diagnóstico y para el mantenimiento de la terapia anticoagulante. También lo es la temperatura de conservación mantenida durante el transporte y almacenamiento de las muestras. Los intervalos de tiempo que se recomiendan entre la obtención de las muestras y la realización de las pruebas son: 2 horas cuando la muestra es mantenida a 22ºC24°C, 4 horas cuando es almacenada a 4°C, 2 semanas a -20°C y 6 meses a - 70°C. Siempre se conserva el tubo tapado hasta su valoración analítica (incluido el tiempo de la centrifugación), para evitar la pérdida de CO2 y la elevación del pH (4). Para la obtención de plasma pobre en plaquetas (PPP), que se utiliza en la mayoría de las pruebas de coagulación, la muestra de sangre se debe centrifugar a 1500 g durante 15 minutos en centrifuga refrigerada 4°C y para obtener el plasma rico en plaquetas (PRP) la muestra se centrifuga a 150–200 g a temperatura ambiente, durante 10 minutos. Se extrae y realizan las pruebas antes de las 4 horas. Los tubos destinados a la investigación del anticoagulante lúpico y la mezcla de plasmas normales, se Revista de Hematología Vol. 7, No. 1, 2006 4 M de los A Ochoa-Rico. aconseja someterlos a doble centrifugación, a 3 000 g, durante 30 minutos (7). FASE ANALÍTICA. Es la etapa que considera a las variaciones relacionadas con el procedimiento técnico en sí mismo, que afectan los resultados finales del estudio del paciente. El control de calidad interno se utiliza para ÿ determinar si una serie de técnicas y procedimientos se están realizando correctamente, durante un determinado periodo de tiempo. Se emplea para asegurar el buen funcionamiento diario del laboratorio (8). Para asegurar que un método está bajo control, a diferentes niveles de un dato concreto, es importante incluir muestras de control de calidad con valores normales y anormales. Los materiales de control de calidad de origen humano tiene las máximas probabilidades de parecerse a las muestras de pruebas humanas. La evaluación interna de la calidad esta basada en el uso de una hoja de control de registró diario (grafica de Levey Jenning) para obtener el control gráfico de la desviación estándar (DE) y calcular el coeficiente de variación (CV), indicadores de precisión de cada tipo de estudio que se realiza a través de un calibrador o control. La gráfica de control de calidad se basa en realizar 20 determinaciones de la prueba a determinar, utilizando un plasma comercial o de fabricación casera de preferencia (mezcla de plasmas). Se representa gráficamente el resultado obtenido del control de cada día de acuerdo a la marca y número de lote del reactivo (figura 1) (7). Preparación del "pool" de plasma. Se recomienda como mínimo 20 personas sanas, que no tomen medicamentos que interferían con los factores y reacciones de la coagulación. Se recolecta la sangre de un número aproximadamente igual de hombres y mujeres. El rango de edad debe estar entre 20 y 50 años, Revista de Hematología Vol. 7, No. 1, 2006 una vez obtenida la sangre colocarla en hielo. Durante la preparación del plasma normal (“pool”), centrifugar a 4°C durante 15 minutos, a 2500 g; mezclar en un contenedor de plástico y poner alícuotas en viales de plástico y congelar inmediatamente en un congelador a -70°C. Hasta 6 meses conservan su estabilidad las muestras (9). ÿ ÿ ÿ ÿ Figura 1.- Curva de Levey – Jenning. El Control de Calidad Externo. Se utiliza para detectar el grado de acuerdo que hay entre los resultados de un laboratorio y los resultados de otros centros. Permite no sólo conocer el funcionamiento de un laboratorio concreto, sino también aquellos reactivos y métodos que producen resultados poco fiables o equívocos. El Comité Internacional de Estandarización en Hematología, ha definido una preparación de referencia como aquella sustancia o instrumento con una o más propiedades suficientemente establecidas como para ser usado para la calibración de un instrumento, para el chequeo de un método de medición o para asignar valores de un material. Existen 3 categorías: estándar primario (internacional), estándar secundario (nacional o regional) y terciario (comercial o local) (7). En las ciencias biomédicas la autoridad en estándares internacionales es la Organización Mundial de la Salud y más recientemente se han incorporado a esta actividad el Comité Internacional 5 Control de calidad en hemostasia. de Estandarización en Hematología, la Federación Internacional de Química Clínica, también el Instituto Nacional para Estándares Biológicos y Control del Reino Unido y el Instituto Nacional para Estándar y Tecnologías del Reino Unido ( 1 0 ) . Estas organizaciones son también responsables de la producción de estándares secundarios. Fase postanalítica. Es la confrontación de todas las fases de análisis y tiene la finalidad de correlacionar los resultados obtenidos con los diagnósticos de los pacientes (11). 9.- Kitchen S, Mc Craw A. Diagnóstico de la hemofilia y otros transtornos de la coagulación. Federanción Mundial de Hhemofilia; 2002. 10.- Koepke JA, Bull BS. The intralaboratory control quality. En: Lewis SM, Koepke JA (eds). Hematology: Laboratory Management and Practice. Oxford: Butterworth-Heinemann; 1995. p. 183-98. 11.- Schman AL, Griner PP. Diagnostic uses of the partial thromboplastin time and prothrombin time. Ann Intern Med 1986; 104:810-6. REFERENCIAS. 1.- Owen CA Jr. Historical account of tests of hemostasis. Am J Clin Pathol 1990; 93:S3-8. 2.- Borzotta AP, Keeling MM. Value of the preoperative history as an indicator of hemostatic disorders. Ann Surg 1984; 200:648-52. 3.- Quintana GS, Martínez-Murillo C, Ambriz FR. Fisiología de la Coagulación, En: Hemofilia, Martínez-Murillo C, Quintana GS, Ambriz FR, Kasper C, editores. México: Editorial Prado; 2001. p. 19-42. 4.- NCCLS. Clinical laboratory procedure manual. Approved guideline. NCCLS Document 10 CP2-A. Villanova, PA:NCCLS, 1984; 4 (2):vii + 27–53. 5.- ICSH. Standardization of blood specimen collection procedure for reference value. Clin Lab Haematol 1982; 4:83-6. 6.- Martínez-Murillo C, Quintana-González S. Fisiología de la Hemostasia Primaria En: Manual de Hemostasia y Trombosis. Martínez-Murillo C, Quintana-González S, editores. México: Editorial Prado; 1996; p. 522. 7.- Lewis SM. Standards, reference materials and reference methods. En: Lewis SM, Koepke JA (eds). Hematology: Laboratory Management and Practice. Oxford: Butterworth-Heinemann;1995. p. 129-35. 8.- Koepke JA, Rodgers H, Ollivier MJ. Preanalytical instrumental variables in coagulation testing. Am J Clin Pathol 1975; 64:591-6. Revista de Hematología Vol. 7, No. 1, 2006 7 Actualización en el diagnóstico y tratamiento de la púrpura trombocitopénica autoinmune. Carlos Martinez-Murillo1,2. 1 2 Servicio de Hematología (Unidad 103). Hospital General de México O.D. Servicio de Hematología y Unidad de Investigación Médica. Hospital General Regional N° 1, Gabriel Mancera. Ciudad de México, D.F., México. INTRODUCCIÓN. La púrpura tromboticopénica autoinmune, también denominada púrpura trombocitopénica inmune o idiopática (PTI) es una enfermedad hemorrágica caracterizada por la destrucción prematura de plaquetas debido a la unión de un autoanticuerpo, habitualmente de la clase IgG, a las glucoproteínas plaquetarias (GPIIb/IIIa) y la posterior depuración del sistema fagocítico mononuclear (1-4). EPIDEMIOLOGÍA. Incidencia. La incidencia general se calcula entre 1 a 12.5 casos (2.25-2.68) por 100,000 personas (5) o bien otras estadísticas informan 100 casos por 1 millón de individuos por año y en niños se informa una incidencia de 4 a 5.3 por 100,000 personas. Estas cifras pueden ser mayores, sin embargo, no existen estudios epidemiológicos que estimen la incidencia real de la enfermedad, incluso muchos casos de PTI aguda en niños no reciben atención médica especializada y no se documentan los casos estadísticos. En niños la prevalencia es la misma entre hombres y mujeres, sin embargo, en adultos la relación mujer-hombre es de 2.6-3:1. En relación a la edad, en los niños la enfermedad no tiene predominio, sin embargo, el pico de prevalencia es de 3 a 5 años. En los adultos la mayor prevalencia se presenta entre los 15 y 40 años. Sin embargo, un estudio realizado en Dinamarca encontró que el promedio de edad fue de 56 años, con un incremento progresivo después de los 60 años (5). Mortalidad y Morbilidad. La primera causa de morbimortalidad de la PTI es la hemorragia. De hecho la hemorragia intracraneal, espontánea o postraumática, constituye la principal causa de muerte cuando la cuenta de plaquetas es menor de 10,000/µL. La mortalidad al momento del diagnóstico es del 1.5%, sin embargo, la mortalidad en pacientes con menos de 30 x 10 9/L plaquetas en los primeros dos años es 4.2 veces mayor (6). La morbilidad asociada al tratamiento puede ser debido a las complicaciones a mediano y largo plazo que provoca el tratamiento de la PTI crónica que es recurrente o resistente al tratamiento, esto ocasionado por el empleo de esteroides, esplenectomía, andrógenos o inmunosupresores (7). Solicitud de reimpresos: Dr. Carlos Martínez-Murillo, Michoacán 18 Casa 1. Col. Miguel Hidalgo, C.P. 14260, México, D.F., México. Tel. 56 06 63 68 E-mail: car1marz@prodigy.net.com Revista de Hematología Vol. 7, No. 1, 2006 8 C Martínez-Murillo. Clasificación. La PTI se clasifica en función del tiempo de evolución en aguda, cuando la duración es menor de 6 meses y crónica cuando esta tiene más de 6 meses de evolución después del diagnóstico. La importancia de determinar si es aguda o crónica, fundamentalmente se asocia con la evolución de la enfermedad. Por ejemplo, en los niños el 70% de los casos son agudos y habitualmente ocurren después de una evento infeccioso y tienen un curso autolimitado (8). En contraste en la población adulta la mayor parte de los casos tienen una evolución a la cronicidad (70 – 80%) (7,9). Se denomina PTI crónica refractaria cuando el paciente no responde a la esplenectomía y mantiene cuenta de plaquetas por debajo de 20-30 x 10 9/L plaquetas y requiere de otras modalidades de tratamiento (10,11). FISIOPATOLOGÍA. Actualmente se conoce que la PTI es mediada por autoanticuerpos. Esto se dedujo de las observaciones en neonatos nacidos de mujeres afectadas con la enfermedad desarrollaban trombocitopenia transitoria. Además estas observaciones fueron confirmadas sobre el fundamento de la presencia de trombocitopenia transitoria en voluntarios sanos, en quienes se les transfundía plasma de individuos afectados con la enfermedad. Así, las plaquetas con los autoanticuerpos IgG tienen una depuración acelerada por el sistema fagocítico mononuclear, a través de los receptores Fcg, que son expresados sobre la superficie de macrófagos tisulares, principalmente de bazo e hígado (1-4). Los autoanticuerpos que reaccionan a las plaquetas, principalmente se unen a las glucoproteínas GPIIb/IIIa, pero también se unen a otros antígenos, como Ib/IX, Ia/IIa, IV y V, así como a otros determinantes antigénicos. De hecho es típica la presencia de anticuerpos contra múltiples antígenos. La destrucción de plaquetas dentro de las Revista de Hematología Vol. 7, No. 1, 2006 células presentadoras de antígenos, puede generar una sucesión de neoantígenos, que resulta en una producción suficiente de autoanticuerpos que ocasiona la trombocitopenia. Los pacientes adultos con PTI tienen linfocitos T HLA DR (+), incremento en el número de receptores para interleucina 2 y un perfil de citocinas que sugieren la activación de precursores de linfocitos T cooperadores y linfocitos T cooperadores tipo 1. En la PTI, las células T estimulan la síntesis de anticuerpos después de la exposición a fragmentos de glucoproteínas IIb/ IIIa. Se desconoce la razón de la expresión de antígenos criptogénicos y de la activación sostenida de linfocitos T. DATOS CLINICOS. Es importante considerar para el diagnóstico de la enfermedad la sintomatología, la evolución de la misma y datos clínicos asociados. La forma aguda de la enfermedad es la presentación característica en los niños en comparación con la presentación crónica de los adultos. La PTI en los adultos tiene un inicio insidioso y habitualmente no le precede una infección viral u otra enfermedad infecciosa. Los signos y síntomas son muy variables y puede ir desde presentaciones asintomaticas hasta pacientes con hemorragias mucocutáneas. Los pacientes con cuenta de plaquetas por arriba de 50 x 10 9/L, el diagnóstico de la PTI es incidental debido a que no presentan sintomatología hemorrágica. Por otro lado, los enfermos con cuenta de plaquetas entre 30 y 50 x 10 9/L tienen petequias y equimosis al mínimo trauma; en contrate los enfermos con cifras de plaquetas de 10 a 30 x 109/L, tienen petequias, equimosis, epistaxis, gingivorragias y/o metrorragias espontáneas. Los pacientes con cifra de plaquetas menor a 10 x 109/L tienen un alto riesgo de hemorragias internas, incluyendo hemorragia en órganos vitales (v. gr. sistema nervioso central) (cuadro 1). 9 Púrpura trombocitopénica autoinmune. Cuadro 1 DATOS CLÍNICOS Petequias Equimosis Gingivorragias Hemorragia transvaginal. Hemorragia conjuntival Hemorragia retiniana Hematuria Hemorragia de tubo digestivo Esplenomegalia Hemorragia en Sístema Nervioso Central (SNC) Datos clínicos presentes en los pacientes con PTI. La presencia de esplenomegalia se puede llegar a observar en el 10% de los pacientes y la hemorragia en SNC se presenta en el 1% (4, 11). DIAGNÓSTICO. El diagnóstico de la PTI todavía sigue efectuándose por exclusión de otros trastornos que ocasionan trombocitopenia. Las presentaciones secundarias pueden asociarse con otros trastornos, como lupus eritematoso generalizado, etc. (cuadro 2). La duración de la hemorragia puede ayudar a distinguir entre la forma aguda y la forma crónica de la PTI. Además, la ausencia de síntomas sistémicos apoya la ruta diagnóstica hacia una PTI primaria. La historia familiar es importante porque distingue entre formas hereditarias como la Púrpura trombocitopénica cíclica familiar de la verdadera PTI (4, 11). Citometría hemática. La citometría hemática se marca la presencia de la cuenta baja de plaquetas y habitualmente el resto de los parámetros hematológicos se encuentran dentro de la normalidad. Sin embargo, en algunos pacientes pueden evidenciarse otros trastornos, como la presencia normocítica normocrómica, que pue- Cuadro 2 Enfermedades asociadas a Trombocitopenia secundaria. Enfermedades Inmunológicas: Lupus eritematoso generalizado. Artritis reumatoide. Síndrome de anticuerpos antifosfolípidos (SAAF). Anemia hemolítica autoinmune (síndrome de Evans). Enfermedades tiroideas autoinmunes Estados de Inmunodeficiencia Síndromes Linfoproliferativos: Leucemia linfocítica crónica Linfomas Infecciones: Virus inmunodeficiencia humana Virus de la hepatitis C Citomegalovirus Asociada a Medicamentos: Heparina Quinidina Antibióticos Trombocitopenia Congénita Asociado a malformaciones esqueléticas. Púrpura trombocitopénica amegacariocítica. de ser secundario a la misma hemorragia o microcítica hipocrómica, asociado a deficiencia de hierro o a padecimiento inflamatorio crónico. En algunos enfermos puede observarse anemia macrocítica que puede estar asociado a datos de deficiencia de hematínicos o la asociación con anemia hemolítica con reticulocitosis. Aspirado de Médula Ósea (AMO). La realización del AMO en los pacientes con PTI ha sido controversial. Sin embargo, las guías publicadas por la Sociedad Americana de Hematología (American Society of Hematology ASH) (7), propone que en adultos de menos de Revista de Hematología Vol. 7, No. 1, 2006 10 C Martínez-Murillo. 60 años, con una presentación típica de la enfermedad, puede evitarse el AMO. Sin embargo, otros autores sugieren que el AMO se realice en individuos de más de 40 años (11). Por otro lado, en los niños se sugiere no realizar AMO si se encuentra bajo vigilancia y tratamiento con altas dosis de inmunoglobulina endovenosa (11), excepto en caso de otras alteraciones hematológicas y presentación atípica. Detección de anticuerpos antiplaquetas. La prueba de inmunoflorescencia es empleada para investigar la presencia de anticuerpos unidos a las plaquetas (PAIgG). El análisis directo de la medición de los anticuerpos unidos a las plaquetas tiene una sensibilidad estimada del 49 al 66%, con una especificidad del 78 al 92% y un valor predictivo positivo del 80 al 83% (4, 11). Por otro lado, la detección de anticuerpos libres en plasma es de menor utilidad, debido a su mayor variabilidad interlaboratorio y su menor sensibilidad y especificidad. La determinación de anticuerpos específicos contra las glucoproteínas plaquetarias GP IIb/IIIa y GPIb/IX son menos sensibles (50 – 65%), pero más específicas (90%). El empleo de estas pruebas pueden ser de utilidad en casos complejos donde existe duda en el diagnóstico y pueden auxiliar a determinar si se trata de una trombocitopenia inmune o no inmune. No deben ser empleadas de rutina. Determinación de trombopoyetina (TPO). Lla determinación de la trombopoyetina puede ser de utilidad en casos complejos, específicamente para distinguir entre trombocitopenia con pobre producción medular (niveles elevados de TPO) o incremento en du destrucción (niveles normales de TPO). Sin embargo, la determinación no debe realizarse como rutina, sólo para casos especiales o para investigación. Helicobacter pylori. Un número de estudios ha informado la presencia de H. pylori en pacientes Revista de Hematología Vol. 7, No. 1, 2006 con PTI y en algunas series la terapia con antibióticos para erradicar el H. pylori ha mejorado casos refractarios de la PTI. A pesar de otros informes contradictorios, puede ser recomendable su determinación en pacientes con PTI crónica refractaria. TRATAMIENTO. Es importante distinguir el criterio para el tratamiento de la PTI, que depende fundamentalmente de la presentación clínica, la cuenta de plaquetas y la evolución de la enfermedad. Los adultos habitualmente requieren tratamiento al inicio de la enfermedad, debido a que la mayoría de ellos se presentan con cuenta de plaquetas por debajo de 50 x 10 9/L. El cuadro 3 muestra las cifras de plaquetas requeridas para la realización de procedimientos invasivos (12). Cuadro 3 Recomendación británica de cifra de plaquetas necesarias para realizar procedimientos. Situación clínica Cifra de plaquetas Tratamiento dental Extracción dental Bloqueo dental regional Cirugía Menor Cirugía Mayor > 10 x 10 9/L > 30 x 10 9/L > 30 x 10 9/L > 50 x 10 9/L > 80 x 10 9/L Tratamiento inicial (primera línea). Corticosteroides.- La primera línea de tratamiento comprende el empleo de corticosteroides, habitualmente prednisona, de 1 a 1.5 mg x kg/día, por 4 a 6 semanas. El porcentaje de respuestas varía del 50 al 75%, con incremento en la cuenta de plaquetas en las primeras dos a tres semanas de tratamiento. Sin embargo, después de la respuesta las recaídas son comunes cuando se reduce la dosis del medicamento y hasta una tercera parte de los pacientes puede tener 11 Púrpura trombocitopénica autoinmune. respuestas prolongadas (10-20%). Pacientes quienes presentan falla a este tratamiento o que requieren de altas dosis para mantener la cuenta de plaquetas segura, deben ser considerados para esplenectomía (12, 13). Inmunoglobulina endovenosa.- La inmunoglobulina endovenosa a altas dosis (IgG AD) es efectiva en elevar la cuenta de plaquetas en el 75% de los pacientes, de los cuales el 50% puede alcanzar cifras normales de plaquetas, sin embargo, produce respuestas transitorias con duración de 3 a 4 semanas y posterior descenso en la cuenta de plaquetas a niveles pre-tratamiento. Un estudio prospectivo no ha demostrado diferencias en la respuesta y necesidad de esplenectomía entre los siguientes tres grupos de tratamiento: 1.- IgG AD; 2.- prednisona o 3.- la combinación. Por otra parte, se ha demostrado que no existe diferencia en los porcentajes de respuesta entre los siguientes esquemas de tratamiento: 0.4 g / kg / día x 5 días y/o 1 g / kg / día (dosis única). El mecanismo de acción involucra varios efectos, como bloqueo de receptores Fc del sistema fagocítico mononuclear, bloqueo en la unión de autoanticuerpos, regulación de la red idiotipo-antiidiotipo y disminución en la producción de autoanticuerpos (13). Falla al tratamiento inicial. Se considera falla al tratamiento de primera línea (corticosteroids y/o IgGAD), a todos aquellos pacientes que requieren altas dosis de corticosteroides para mantener una cifra segura de plaquetas. El porcentaje de estos fluctúan entre 11% y 35% (14). Otro aspecto a considerar, es la necesidad de mantener por tiempo prolongado el empleo de corticosteroides a altas dosis, por los efectos adversos que ocasiona a los enfermos. De tal suerte que en caso de falla se debe considerar realizar la siguiente opción terapéutica de elección, como la esplenectomía. Sin embargo, en ese lapso que el pacien- te se esplenectomiza, debe mantenerse un nivel seguro de plaquetas mediante una dosis de esteroides y la combinación de otro fármaco. Un gran número de medicamentos han sido empleados para mantener una cifra estable de plaquetas. Esta terapia depende de la edad del enfermo, la gravedad de la presentación, el nivel de plaquetas, tiempo de evolución, estado general del enfermos, etc. Entre las alternativas de tratamiento existen las siguientes opciones; IgG AD, IgG anti-D, danazol, alcaloides de la vinca (cada vez en menor empleo por la pobre respuesta), etc. Tratamiento de segunda línea. Esplenectomía. La esplenectomía permanece aún como la segunda línea de tratamiento cuando han fallado medidas terapéuticas previas. El procedimiento no es estrictamente “curativo”, debido a que el mecanismo inmunológico persiste y únicamente se remueve uno de los principales sitios de destrucción. Hasta el 70% de los pacientes puede tener respuesta y alcanzar cifras normales de plaquetas. Algunos pacientes pueden tener respuestas tardías después del procedimiento. A. Recomendaciones pre-operatorias. Se recomienda que a los pacientes se les incrementa los niveles de plaquetas a base de IgG AD, prednisona o bolos de dexametasona, con objeto de disminuir los riesgos de hemorragia perioperatoria. Stasi y col. (15) recomiendan tratamiento para pacientes con cuenta de plaquetas por debajo de 30 x 109/L. Es importante la prevención de la infección post-esplenectomía principalmente contra pneumococo, aunque también existen vacunas para prevenir la infección por Haemophilus influenzae y meningococo (vacunas conjugadas), para lo cual es necesaria la administración dos semanas previas a la cirugía de la vacuna (ejem. Pneumovax) y administrarla cada cinco años. En pacientes esplenectomizados se Revista de Hematología Vol. 7, No. 1, 2006 12 C Martínez-Murillo. recomienda la vacunación anual de la vacuna contra influenza. Algunos estudios recomiendan el uso de penicilina o eritromicina en los primeros 3 años después de la esplenectomía, sin embargo, no hay estudios que hayan documentado su eficacia en la prevención de la infección por neumococo. Sin embargo, es importante que el paciente siempre lo mencione en caso de alguna urgencia u hospitalización. B. Soporte Operatorio. Aunque el procedimiento es sencillo requiere que el grupo quirúrgico tenga experiencia en este tipo de enfermos. Se ha sugerido que el paciente requiere únicamente soporte transfusional con plaquetas únicamente mientras se liga la arteria esplénica. Sin embargo, no existen estudios sólidos a este respecto. C. Cuidados post-operatorios. Las medidas postoperatorias implican vigilancia de complicaciones asociadas al procedimiento (22%), entre las que incluyen embolismo pulmonar, absceso abdominal, hematoma de la pared abdominal, sepsis y otras. Otros estudios han informado 0% de mortalidad y 7 % de morbilidad. D. Búsqueda de bazo accesorio. La presencia de un bazo accesorio debe sospecharse en aquellos pacientes que tienen falla a la esplenectomía o que recaen después de una respuesta inicial. La mejoría en las técnicas radiológicas ha permitido detectar estos bazos hasta en el 12% de estos pacientes. E.- Predictores de la respuesta a la esplenectomía. El mejor predictor de la respuesta a la esplenectomía es el estudio de plaquetas autólogas marcadas con indium, de acuerdo con el estudio de Najean y cols(16) en 528 pacientes. Los pacientes donde existe destrucción esplénica, > 90% pueden obtener remisión. En contraste los pacientes con destrucción plaquetaria hepática o mixta Revista de Hematología Vol. 7, No. 1, 2006 (hepática y esplénica), el 92% tuvieron falla a la esplenectomía. Falla a la Esplenectomía. Se considera falla a la esplenectomía a todos aquellos pacientes que posterior al procedimiento presentan cuenta de plaquetas <50 x 10 9/L. En este caso se denomina PTI Crónica Refractaria y requiere de múltiples opciones de tratamiento. A. Corticosteroides.- En caso de falla a la esplenectomía se recomienda el empleo de corticosteroides a altas dosis para intentar una remisión completa y entre las formas de empleo se encuentra la convencional, con prednisona de 1 a 1.5 mg x kg/día, por 2 a 4 semanas, o bien el esquema de altas dosis de dexametasona (17, 18), que consiste en 40 mg/día, por 4 días, cada 28 días hasta completar 6 ciclos. También puede ser empleado el esquema de bolos de metil prednisolona con el siguiente esquema: 30 mg/kg/ día por 3 días, seguido de 20 mg /kg/ día por 4 días y después 5, 2 y 1 mg/kg/ día por 1 semana. La respuesta habitualmente se observa dentro de los primeros 3 a 5 días (19). B. Altas dosis de Ig.- El empleo de Ig AD a la dosis de 1 g/kg/día, por 2 días consecutivos, en asociación con corticosteroides, incrementa rápidamente la cuenta de plaquetas (20, 21) y resulta mejor cuando se repite cada tres semanas (22), donde se informa de remisiones completas. Sin embargo, en general, las respuestas con Ig AD son transitorias y rara vez produce respuestas prolongadas (22-24), por lo tanto este tipo de tratamiento habitualmente esta reservado para pacientes sintomáticos. C. Inmunoglobulina anti-D.- Desde hace varios años se ha empleado la IgG anti-D para el tratamiento de la PTI. Salama y col. (25, 26) identificaron que la infusión de Ig AD en pacien- 13 Púrpura trombocitopénica autoinmune. tes con PTI fue asociado con evidencia de laboratorio de hemolisis. Entonces hipotetizó que en las preparaciones de inmunoglobulinas existían pequeñas cantidades de anticuerpos anti eritrocitos que podrían ser los responsables del bloqueo al receptor Fc (FcR) y por lo tanto del incremento en la cifra de plaquetas. Con el objetivo de probar esta hipótesis, administraron IgG anti-D en pacientes con PTI Rh (D+) y obtuvieron incremento en la cuenta de plaquetas en la mayoría de los enfermos. Posteriormente otros autores han documentado el mismo efecto (27-35). Scaradavou y col. (36) informaron que entre 79 y 90% de los adultos que fueron tratados con IgG anti-D tuvieron respuesta. Eritrocitos Opsonizados con IgG anti-D. Desde 1984 (38) se ha publicado la experiencia con el empleo de IgG anti-D en forma de eritrocitos autólogos opsonizados para el tratamiento de la PTI crónica refractaria, donde se han obtenido respuestas de más del 60% (3842). Estas respuestas han sido también obtenidas por Ruíz-Arguelles y col. (43) con la misma forma de tratamiento. D. Danazol.- El danazol es un andrógeno sintético, con pocos efectos virilizantes, que ha sido empleado de manera sinérgica con los corticosteroides. Ahn y col. (44) informó que de 22 pacientes tratados con danazol a las dosis de 200 mg 2 a 4 veces al día, por más de 2 meses, el 60% presentó incremento en la cuenta de plaquetas por más de 2 meses. El mecanismo de acción es desconocido, pero parece ser que disminuye la expresión de receptores Fc sobre el sistema fagocítico mononuclear del bazo. E. Inmunosupresores.- La inmunosupresión puede ser requerida si fallan los tratamientos previos. El tratamiento con azatioprina 2 mg/kg/ día (máximo 150 mgs) o ciclofosfamida produce respuestas mayores al 25% y la mayoría de ellas sostenidas (45). Por su parte, Quiquandon y col. (46) informaron que de 53 pacientes tratados con azatioprina por una media de 18 meses, 64% tuvieron incremento en la cuenta de plaquetas y en 45% tuvo remisiones completas. La azatioprina tiene un efecto lento y debe ser administrado por un mínimo de 6 meses, antes de considerar que existe falla al tratamiento. F. Dapsona.- La dapsona es un fármaco tradicionalmente empleado para la lepra. Sin embargo, en algunos adultos con PTI crónica tratados con este medicamento, a la dosis de 75 a 10 mgs/día, por 21 días, se obtuvo 50% de respuestas (47). El mecanismo de acción de la dapsona es desconocido sin embargo, puede ser debido al bloqueo del sistema fagocítico mononuclear a través del incremento en la destrucción de glóbulos rojos (48). Parece ser que las mejores respuestas a este tratamiento es en casos no graves y que no han sido esplenectomizados. G. Anticuerpos monoclonales.- El anti CD20, denominado rituximab ha sido evaluado en algunas series de pacientes con PTI crónica refractaria a la dosis de 375mg/m2 sc, semanalmente por 4 semanas, en los cuales se observó algún tipo de respuesta en el 50% (la mitad de ellos con remisión completa), con respuestas de más de 6 meses. Existe la sugerencia que los pacientes jóvenes responden mejor al tratamiento (49). El otro anticuerpo monoclonal que se ha empleado es el anti-CD52, denominado alemtuximab (CAMPATH), el cual ha sido aprobado para el tratamiento de la leucemia linfocítica crónica y que se ha empleado en pacientes con pancitopenias inmunológicas, incluyendo algunos pacientes con PTI crónica refractaria, en los cuales se han informado respuestas después de 3 a 4 semanas de tratamiento, con respuestas sostenidas de más de 4 a 9 meses. Sin embargo, el efecto adverso más importante es la inmunodepresión con Revista de Hematología Vol. 7, No. 1, 2006 14 C Martínez-Murillo. linfopenia severa (< 0.110 9 / L) y en algunos pacientes se ha informado el agravamiento de la trombocitopenia (50). H. Micofenolato de Mofetil.- Este inmunosupresor antiproliferativo ha sido aprobado para la prevención del rechazo agudo en pacientes trasplantado. Ha demostrado eficacia en el tratamiento de la PTI crónica refractaria, pero requiere de mayor comprobación en un mayor número de enfermos (51). I. Otras opciones de tratamiento.Considerando el riesgo beneficio de otras modalidades de tratamiento, tales como interferón alfa, inmunoadsorción con columnas de proteína A, plasmaféresis, doxorrubicina liposomal y alcaloides de la vinca, estas opciones terapéuticas ahora no son recomendadas (11). EVOLUCIÓN. La edad media de la presentación fue a los 39 años y la mayoría de los pacientes presentan trombocitopenia severa. Durante los primeros 2 años alunos pacientes pueden manifestar las siguientes enfermedades; lupus eritematoso generalizado, artritis reumatoide, síndrome de anticuerpos antifosfolípidos, colitis crónica, linfomas u otros tipos de cáncer, etc. REFERENCIAS. 1.- Karpatkin S. Autoimmune thrombocytopenic purpura. Semin Hematol 1985; 22: 260-88. 2.- Waters AH. Autoimmune thrombocytopenia: Clinical Aspects. Semin Hematol 1992; 29: 18-25. 3.- George JN, Raskob GE. Idiopathic thrombocytopenia purpura: A concise summary of the pathophysiology and diagnosis in children and adults. Semin Hematol 1998; 36: 5-8. 4.- Cines DB, Blanchette V. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995-1008. Revista de Hematología Vol. 7, No. 1, 2006 5.- Henrik-Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood 1999; 94: 909-13. 6.- Johanna E, Portielje A, Rudi G, Westendorp J, Hanneke C. Kluin-Nelemans, et al. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 9:2549-54. 7.- George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood 1996; 88: 3-40. 8.- Calpin C, Dick P, Foon A, Feldman W. Is bone marrow aspiration needed in acute childhood idiopathic thrombocytopenic purpura to rule out leukemia? Arch Pediatr Adolesc Med 1998; 152:345-7. 9.- Chong BH, Keng TB. Advances in the diagnosis of idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:249-60. 1 0 . - Ta r a n t i n o M . A c u t e I m m u n e ( i d i o p a t h i c ) thrombocytopenic purpura in childhood. Blood Rev 2002; 16:19-21. 11.- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol, 2003; 120:574–96. 12.- Pizzuto J, Ambriz R. Therapeutic experience on 934 adults with idiopathic thrombocytopenic purpura: Multicentric Trial of the Cooperative Latin American group on Hemostasis and Thrombosis. Blood 1984; 64:1179–83. 13.- Warkentin TE, Kelton JG. Current concepts in the treatment of immune thrombocytopenia. Drugs 1990; 40:531–42. 14.- George JN, el-Harake MA, Raskob GE. Chronic idiopathic thrombocytopenic purpura. N Engl J Med 1994; 331:1207–11. 15.- Stasi R, Stipa E, Masi M, Cecconi M, Scimo MT, Oliva F, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995; 98:436–42. 15 Púrpura trombocitopénica autoinmune. 16.- Najean Y, Rain JD, Billotey C. The site of destruction of autologous 111In-labelled platelets and the efficiency of splenectomy in children and adults with idiopathic thrombocytopenic purpura: a study of 578 patients with 268 splenectomies. Br J Haematol 1997; 97: 547–50. 17.- Andersen JC. Response of resistant idiopathic thrombocytopenic purpura to pulsed high-dose dexamethasone therapy. N Engl J Med 1994; 330:1560– 64. 18.- Arruda VR, Annichino-Bizzacchi JM. High-dose dexamethasone therapy in chronic idiopathic thrombocytopenic purpura. Ann Hematol 1996; 73: 175–7. 19.- Akoglu T, Paydas S, Bayik M, Lawrence R, Firatli T. Megadose methylprednisolone pulse therapy in adult idiopathic thrombocytopenic purpura. Lancet 1991; 337: 56. 20.- Bussel JB, Hilgartner MW. The use and mechanism of action of intravenous immunoglobulin in the treatment of immune haematologic disease. Br J Haematol 1984; 56: 1–7. 21.- Imbach P, Wagner HP, Berchtold W, Gaedicke G, Hirt A, Joller P, et al. Intravenous immunoglobulin versus oral corticosteroids in acute immune thrombocytopenic purpura in childhood. Lancet 1985; 2: 464–8. 22.- Schiavotto C, Ruggeri M, Rodeghiero F. Adverse reactions after high-dose intravenous immunoglobulin: incidence in 83 patients treated for idiopathic thrombocytopenic purpura (ITP) and review of the literature. Haematologica 1993; 78: 35–40. 23.- Schiavotto C, Ruggeri M, Rodeghiero F. Failure of repeated courses of high-dose intravenous immunoglobulin to induce stable remission in patients with chronic idiopathic thrombocytopenic purpura. Ann Hematol 1995; 70:89–90. 24.- Bussel J. Novel approaches to refractory immune thrombocytopenic purpura. Blood Rev 2002; 16:31-6. 25.- Salama A, Kiefel V, Amberg R, Müeller-Eckhardt C. Treatment of autoimmune thrombocytopenic purpura with rhesus antibodies (anti-RhO [D]). Blut 1984; 49: 29-35. 26.- Salama A, Kiefel V, Mueller--Eckhardt C. Effect of IgG anti-Rho(D) in adult patients with chronic autoimmune thrombocytopenia. Am J Hematol 1986; 22: 241-50. 27.- Baglin TP,Smith MP, Boughton BJ. Rapid and complete response of immune thrombocytopenic purpura to a single injection of rhesus anti-D immunoglobulin. Lancet 1986; 1: 1329-30. 28.- Panzer S, Grumayer ER, Haas OA, Niesner H, Graninger W. Efficacy of rhesus antibodies (Anti-Rho (D)) in autoimmune thrombocytopenia: Correlation with response to high dose IgG and the degree of haemolysis. Blut 1986; 52: 117-21. 29.- Gabra GS, Mitchell R. Anti-D immunoglobulin and immune thrombocytopenia- a problem of ethics in blood transfusion practice. Vox Sang 1988; 54: 246. 30.- Bussel JB, Graziano JN, Kimberly RP, Pahwa S, Aledort LM. Intravenous anti-D treatment of immune thrombocytopenic purpura: Analysis of efficacy, toxicity and mechanism of effect. Blood 1991; 77: 188493. 31.- Ware RE, Zimmerman S. Anti-D: Mechanisms of action. Semin Hematol 1998; 36: 14-22. 3 2 . - G e o rg e J N , E l - H a r a k e M A , A s t e r R H . Thrombocytopenia due to enhance platelet destruction by immunologic mechanisms. Williams Hematology 5th ed. New York: Mc Graw-Hill, Inc. 1995. p. 1315-55. 33.- Bussel JB. Recent advances in the treatment of idiopathic thrombocytopenic purpura: the anti-D clinical experience. Semin Hematol 1998; 36: 1-4. 34.- Borgna-Pignatti C, Battisti L, Zecca M, Locatelli F. Tr e a t m e n t o f c h r o n i c c h i l d h o o d i m m u n e thrombocytopenic purpura with intramuscular anti-D immunoglobulins. Br J Haematol 1994; 88: 618-20. 35.- Bierling P, Karianakis G, Duedari N, Desaint C, Oksenhendler E, Habibi H. Anti-Rhesus antibodies immune thrombocytopenia and human immunodeficiency virus infection. Ann Intern Med 1987; 106: 773-4. 36.- Scaradavou A, Woo B, Woloski, BM, CunninghamRundles S, Ettinger LJ, Aledort LM, Bussel JB. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Revista de Hematología Vol. 7, No. 1, 2006 16 C Martínez-Murillo. Blood 1997; 89: 2689–700. 37.- Ambriz R, Pizzuto J, Morales M, Muñoz R, Quintanar G, García N. Treatment of chronic ITP with opsonized erythrocytes with rhesus antibody and labeled with Tc-99m (Radioimmune method). Blood 1984; 64:(suppl):233. 38.- Ambriz R, Quintanar E, Domínguez JL, García EN, Estrada C, Alvarez E, et al. Tratamiento con IgG anti D (Rho) en PTA refractaria, mediante transfusión directa (TD) vs eritrocitos opsonizados "in vitro" (TEOP), Trabajo Multicéntrico AMEH, A.C. Sangre 1987; 32:259. 39.- Ambriz R, Muñoz R, Quintanar E, Sigler L, Avilés A, Pizzuto J. Accessory spleen compromising response to splenectomy for idiopathic thrombocytopenic purpura. Radiology 1985; 155: 793-796. 40.- Ambriz R, Muñoz R, Quintanar E, Sigler L, Avilés A, Pizzuto J. Accessory spleen compromising response to splenectomy for idiopathic thrombocytopenic purpura. The Year Book of Nuclear Medicine. Chicago: Year Book Medical Publishers Inc; 1987. p. 326-7. 41.- Ambriz R, Muñoz R, Pizzuto J, Quintanar E, Morales M, Avilés A. Low-dose autologous in vitro opsonized erythrocytes. Radioimmune method and autologous opsonized erythrocytes for refractory autoimmune thrombocytopenic purpura in adults. Arch Intern Med 1987; 147: 105-8. 42.- Ambriz FR, Martínez-Murillo C, Quintana GS, Collazo-Jaloma J, Bautista JJ. Fc Receptor blockade in patients with refractory chronic immune thrombocytopenic purpura. Arch Med Research 2002; 33: 536-40. 43. Ruíz-Argüelles GJ, Apreza MMG, Perez-Romano B, Ruiz-Argüelles A. The infusion of Anti-Rho (D) opsonized erythrocytes may be useful in the treatment of patients, splenectomized or not, with chronic, refractory autoimmune thrombocytopenic purpura. A prospective study. Am J Hematol 1993; 43: 72-3. 44.- Ahn YS. Rocha R. Mylvaganam R. Garcia R, Duncan R, Harrington WJ. Long-term danazol therapy in autoimmune thrombocytopenia: unmaintained remission and agedependent response in women. Ann Intern Med 1989; 111:723–9. Revista de Hematología Vol. 7, No. 1, 2006 45.- Bouroncle BA, Doan CA. Treatment of refractory idiopathic thrombocytopenic purpura. JAMA 1969; 207: 2049–2052. 46.- Quiquandon I, Fenaux P, Caulier MT, Pagniez D, Huart JJ, Bauters F. Re-evaluation of the role of azathioprine in the treatment of adult chronic idiopathic thrombocytopenic purpura. a report on 53 cases. Br J Haematol 1990; 74: 223–8. 47.- Godeau B, Durand JM, Roudot-Thoraval F, Tenneze A, Oksenhendler E, Kaplanski G, et al. Dapsone for chronic autoimmune thrombocytopenic purpura: a report of 66 cases. Br J Haematol 1997; 97:336-9. 48.- Radaelli F, Calori R, Goldaniga M, Guggiari E, Luciano A. Adult refractory chronic idiopathic thrombocytopenic purpura: can dapsone be proposed as second-line therapy? [Letter]. Br J Haematology 1999; 104:641–2. 49.- Stasi R, Pagano A, Stipa E, Amadori S. Rituximab chimaeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood 2001; 98:952–7. 50.- Willis F, Marsh JC, Bevan DH, Killick SB, Lucas G, Griffiths R, Ouwehand W, Hale G, Waldmann H, GordonSmith EC. The effect of treatment with Campath-1H in patients with autoimmune cytopenias. Br J Haematol 2001; 114:891–8. 51.- Howard J, Hoffbrand AV, Prentice HG, Mehta A. Mycophenolate mofetil for the treatment of refractory autoimmune haemolytic anaemia and auto-immune thrombocytopenia purpura. Br J Haematology 2002; 117: 712–5. 17 Tratamiento de la enfermedad de von Willebrand. Sandra Quintana-González 1, Carlos Martínez-Murillo 2, 3. 1 Banco Central de Sangre, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, 2Servicio de Hematología (Unidad 103). Hospital General de México O.D., 3Servicio de Hematología y Unidad de Investigación Médica. Hospital General Regional N° 1, Gabriel Mancera. Ciudad de México, D.F., México. INTRODUCCIÓN. La enfermedad de von Willebrand (EvW) es una enfermedad hemorrágica autosómica hereditaria causada por la deficiencia o disfunción del factor de von Wilebrand (FvW), que se caracteriza por hemorragias mucocutáneas de intensidad variable y que afecta primordialmente la hemostasia primaria en la interacción plaqueta, FvW y endotelio. El FvW es una proteína multimérica que tiene dos funciones en la hemostasia. Es esencial para la formación del coágulo plaquetario por sus funciones en la adhesión y agregación plaquetaria, a través de los grandes multímeros del factor, y la formación de un complejo con el factor VIII por medio de una unión no covalente, protegiendo a este factor de la degradación enzimática. Por lo tanto, contribuye indirectamente al proceso de coagulación o hemostasia secundaria. Este defecto hemorrágico de origen genético se codifica en el cromosoma 12, cromosoma que se encarga de codificar la información para una molécula madura, con una gran heterogeneidad, que produce variaciones biológicas en la enfermedad. En 1926, Erik von Willebrand describió una enfermedad hemorrágica en una familia numerosa en las Islas Aland, en el golfo de Botnia, en las costas de Finlandia (1). A diferencia de la hemofilia, en esta enfermedad uno y otro sexo eran afectados y la hemorragia mucocutánea predominaba. Erik von Willebrand asignó el término de “pseudohemofilia hereditaria” para designar al padecimiento que tenía como característica común, hemorragias mucocutáneas de intensidad variable, con herencia autosómica y tiempo de hemorragia (TH) prolongado. Posteriormente Jürgens colabora con Erik von Willebrand y al estudiar a los enfermos de las islas Aland consideran el término de “Trombopatía constitucional von Willebrand-Jürgens” por considerar que se trataba de un defecto plaquetario. En 1957, se informó que el defecto podía ser corregido por un factor plasmático diferente al factor VIII (FVIII), denominándose factor de von Willebrand (FvW) (2). La reducción del FvW causa reducción del Factor VIII, observando la estrecha relación que tienen ambas proteínas. La purificación del FvW y el subsecuente desarrollo de reactivos serológicos y técnicas electroforéticas especializadas, han permitido conocer la heterogenicidad del FvW (3-6). Solicitud de reimpresos: Dra. Sandra Quintana-González, Aureliano Rivera No. 1 casa 8, Tizapan San Ángel, C.P. 01090, México, D.F., México. Tel. 56 45 86 13 E-Mail: sanquin@prodigy.net.mx Revista de Hematología Vol. 7, No. 1, 2006 18 S Quintana-González, C Martínez-Murillo. INCIDENCIA. La EvW es la enfermedad hemorrágica hereditaria más frecuente, con una distribución mundial y sin predominio de sexo. Se ha informado una prevalencia del 0.9%, aproximadamente 8.2 casos por 1000 habitantes y se ha determinado una prevalencia del 1.3% en población multiétnica (7). De los pacientes con EvW el 70 al 80% son tipo 1, 5 al 15% tienen alguna variedad del tipo 2 y la prevalencia del tipo 3 (EvW severa) es de 1 a 5 por millón de habitantes en Europa y de 3 por millón en Suecia e Israel. En Alemania el tipo 3 representa el 12% de los casos, en Italia el 17% y en Israel hasta el 29% (8-10). En Latinoamérica se ha informado una incidencia de 1.1% en Costa Rica (11). FACTOR DE VON WILLEBRAND (FvW). El FvW es una glucoproteína de alto peso m o l e c u l a r si ntetizado y alm acena do e n megacariocitos y células endoteliales. El gene que codifica el FvW ha sido clonado y localizado en el cromosoma 12p13.2. El gene está compuesto de 178 kilobases con 52 exones. La estructura del FvW está compuesta de un polipéptido de 270 kD, con una subunidad que comprende 2,050 residuos de aminoácidos; cada subunidad contiene sitios de unión para la colágena y para las glicoproteínas (Gp) Ib y GpIIb/IIIa (fig.1). En vasos sanguíneos intactos el FvW no interactúa con los receptores de plaquetas. Cuando el vaso se daña expone el subendotelio y se une el FvW. Esta interacción induce un cambio conformacional en el FvW, que expone los sitios de unión para que la GpIb de las plaquetas se una al FvW y se lleve a cabo el mecanismo de adhesión plaquetaria por medio del dominio A1 . El FvW se adhiere a la fibras de colágena de la pared vascular, pero también a otros componentes del subendotelio (12). Por otro lado, en superficies con “high shear stress” se ha demostrado la activación del sitio de unión de la GpIIb/IIIa (IIb3) sobre la membrana plaquetaria. Esta activación es capaz de unir plaquetas (agregación) por medio del FvW, fibrinógeno, vitronectina y otras proteínas que contengan la secuencia Arg-Gly-Asp. El ARN m codifica para una proteína de alrededor de alrededor de 2,813 aminoácidos (aa) llamada pre-pro-FvW. Este producto inicial de 300 a 350 Kd pierde una fracción llamada “péptido de señal” (SP), que consta de 22 aa, que inicia el proceso de formación de la proteína del FvW (fig 2a). Después de esta pérdida el pro-polipéptido de 2791 aa (fig 2b), forma dímeros a través de la formación de puentes disulfuro en las porciones carboxi-terminales (fig 2c). Posteriormente se lleva a cabo la glucosilación en el aparato de Golgi, lo que α β Fig.1 Estructura del Pro-factor de von Willebrand (FvW). En la figura se señala la organización de los dominios del FvW. Estos dominios son definidos y agrupados de acuerdo a su homología interna. Las barras negras indican la localización de los sitios de unión. La secuencia en el dominio C1 interviene en la unión de la GpIIb/ IIIa (IIb/3), pero el estado funcional del dominio D2 permanece desconocida. La unión S-S indica la localización de los puentes disulfuro involucradas en la dimerización y multimerización. Revista de Hematología Vol. 7, No. 1, 2006 19 Enfermedad de von Willebrand. Figura 2.- Secuencia multimérica del factor de von Willebrand (FvW). resulta en un alto contenido de carbohidratos. La proteína madura de 2051aa, forma puentes de disulfuro en las porciones amino terminales de los dímeros, se forman series de multímeros de diferente tamaño que van desde una sola unidad fundamental de 225 kd hasta 120, 000 Kd (13) (fig 2d). Los multímeros del FvW se almacenan en su mayor parte en los cuerpos de Weibel Palade del endotelio y bajo ciertos estímulos pasan a circulación y al subendotelio. Los productos que liberan al FvW son: trombina, calcio, fibrina, Revista de Hematología Vol. 7, No. 1, 2006 20 S Quintana-González, C Martínez-Murillo. Figura 3.- Complejo factor VIII/factor de von Willebrand. El factor VIII se une a la proteína multimérica del factor de von Willebrand por medio de una unión no covalente. activador tisular del plasminógeno (t-PA), plasmina, adrenalina bradicinina, interleucina-1, vasopresina y su análogo sintético, la desaminoD-arginina-vasopresina (DDAVP), o del FvW y favorecen su actividad biológica. El FvW funciona como el acarreador esencial del FVIII permitiendo la estabilidad de este factor en la circulación. El FVIII circula en plasma con el factor de von Willebrand (FvW) para evitar que el factor VIII, el cual es lábil se destruya. Por lo tanto, el FvW es la molécula que protege al FVIII de la destrucción de algunas enzimas en plasma y es el factor que le da estabilidad al factor VIII. La unión del factor VIII con el FvW es no covalente y recibe el nombre de Complejo FVIII:C/FvW, el cual es un complejo estable (fig.3). El FvW se une a la GPIb-IX y establece el contacto inicial entre las plaquetas y la superficie subendotelial (colágena), es decir favorece los mecanismos de adhesión plaquetaria. Esto ocasiona la activación primaria de la plaqueta. La activación plaquetaria ocasiona la liberación de productos almacenados en los gránulos alfa y cuerpos densos, incluyendo FvW plaquetario y el cambio conformacional de la GPIIb-IIIa. El FvW se une a la GPIIb-IIIa y participa en los mecanismos de interacción plaqueta-plaqueta, Revista de Hematología Vol. 7, No. 1, 2006 mediante el mecanismo de agregación plaquetaria, donde participa el fibrinógeno y iones de calcio. En la figura 4 se representa de manera esquemática los mecanismos de adhesión plaquetaria. El primer contacto se establece entre las plaquetas y el FvW por medio del receptor glucoprotéico Ib. Esta unión se realiza a través del dominio A1 del FvW. Las plaquetas rápidamente pueden unirse a las superficies cubiertas con el FvW, siempre que existan condiciones de flujo y deben tener también alta resistencia a la fuerza de tracción. Después de que las plaquetas se adhieren pueden resistir a la fuerza creada por el flujo que resulta en las paredes con cizallamiento. La interacción, tiene una elevada velocidad de disociación intrínseca, resultando en una rápida separación por el movimiento de rotación impuesto por el flujo sanguíneo. Se forman nuevas uniones en diferentes regiones de la membrana de las plaquetas en rotación, en estrecho contacto con la superficie. La translocación continúa hasta que el receptor GpIIb/IIIa, que inicialmente no se puede unir con el FvW, posteriormente se activa y se une a la secuencia RGDS del dominio C1 del FvW. Finalmente, se unirán otras plaquetas a la superficie produciendo el fenómeno de agregación plaquetaria. 21 Enfermedad de von Willebrand. Figura 4.- Adhesión plaquetaria al factor de von Willebrand. En la pared vascular intacta, el flujo sanguíneo provoca que los eritrocitos y los leucocitos se encuentren en el centro del vaso sanguíneo y las plaquetas se encuentran más cercanas a la pared vascular. Sin embargo, las células endoteliales impiden la interacción de estas plaquetas a la pared intacta del vaso, ya que las fibras de colágena se encuentran en la matriz subendotelial. Cuando la pared vascular esta intacta y el flujo sanguíneo es normal, el FvW que circula en el plasma y las plaquetas pueden tener mínimas interacciones. En la pared vascular dañada, las fibras de colágena y el FvW se exponen al flujo sanguíneo y a las fuerzas de cizallamiento. El FvW plasmático eficientemente se une a la colágena expuesta por medio de la GpIa y su estructura se desenrolla, apoyando la adhesión de las plaquetas circulantes en sinergia con la colágena. La unión del FvW interactúa primero solamente con el receptor GpIb e inicia la rotación de las plaquetas (fig. 4 y 5b). Esta interacción se disocia rápidamente y la rotación de las plaquetas se realiza de acuerdo al flujo sanguíneo. Una vez que las plaquetas están activadas se forman pseudópodos incrementando la afinidad del factor de von Willebrand y el receptor GpIIb/IIIa se activa y presenta un cambio conformacional en la superficie de las plaquetas, lo que ayuda a la interacción de plaqueta-plaqueta (agregación), formando una coágulo plaquetario a través del FvW y a las condiciones bajas del flujo sanguíneo y al fibrinógeno (fig. 5c). CLASIFICACIÓN. La identificación de varios subtipos de la EvW ha contribuido a su complejidad, además de las variaciones en la herencia, manifestaciones clínicas y resultados de las pruebas de hemostasia. El tratamiento de la EvW depende en gran medida del subtipo de la enfermedad. Los progresos recientes en la caracterización de las mutaciones que causan la EvW, han proporcionado datos suficientes para reorganizar la forma como había sido históricamente clasificada la enfermedad. En 1994 se publicó un nuevo sistema de clasificación para la EvW. Está basada principalmente en el fenotipo de la proteína del FvW, que está presente en el plasma y plaquetas del paciente (6). La clasificación identifica dos categorías por alteraciones cuantitativas del FvW (Tipos 1 y 3) o por alteraciones cualitativas del FvW (Tipo 2). La deficiencia cuantitativa del FvW en plasma y/o plaquetas identifica a la EvW tipo 1, mientras que la EvW tipo 3 se encuentra ausente Revista de Hematología Vol. 7, No. 1, 2006 22 S Quintana-González, C Martínez-Murillo. Figura 5.- Función del factor de von Willebrand en hemostasia primaria. o solamente pequeñas cantidades de FvW en plasma y plaquetas se encuentran presentes. El tipo 1 se diferencia del tipo 3 por la deficiencia leve del FvW (usualmente de 30-40 UI/dL), la herencia autosómica dominante y la presencia de hemorragias leves. Se identifican cuatro subtipos de la EvW tipo 2. Estos reflejan los mecanismos fisiopatológicos distintos entre cada uno de ellos. El tipo 2A y 2B se caracterizan por la ausencia de los multímeros de gran tamaño en el plasma; en el tipo 2B, existe un aumento de la afinidad del FvW a la GpIbα. La identificación de las variantes cualitativamente anormales del FvW con disminución de la función dependiente de Revista de Hematología Vol. 7, No. 1, 2006 plaquetas y la presencia de multímeros normales, ha caracterizado al subtipo 2M, causado por mutaciones que afectan la función del FvW, pero no afectan la estructura multimérica. En el tipo 2N (Normandy), la estructura multimérica del FvW no está alterada, sin embargo la región Nterminal sobre el FvW no se une al Factor VIII, por lo que solamente se puede identificar por la prueba de unión del FvW/FVIII. CUADRO CLÍNICO. Clínicamente la enfermedad se caracteriza por la presencia de hemorragias mucocutáneas de intensidad variable y que tiende a ser fluctuante, es decir alternan períodos 23 Enfermedad de von Willebrand. hemorrágicos con períodos asintomáticos, lo que dificulta el diagnóstico de la enfermedad. Los síntomas son más intensos en los niños y adolescentes. Además, gran variación en la frecuencia y severidad de la enfermedad existen dentro de las familias afectadas. La expresión clínica de la EvW usualmente es leve en el tipo I y la severidad aumenta en los tipos 2 y 3. En general, la severidad de la hemorragia correlaciona con el grado de reducción del FVIII:C pero no con el TH. La epistaxis el principal síntoma en estos pacientes, con una frecuencia del 60%; las metrorragias constituyen el principal síntoma en las mujeres adolescentes, cuya frecuencia puede alcanzar cifras hasta del 75% (cuadro 1). Los niños frecuentemente presentan equimosis de aparición espontánea, que sugiere la posibilidad de EvW. Por otra parte, la EvW puede ser diagnosticada después de un procedimiento quirúrgico con hemorragia transoperatoria y postoperatoria, particularmente después de e xtracciones dentales o amigdalectomía. Usualmente, el factor VIII se encuentra discretamente disminuido, por lo tanto, las manifestaciones hemorrágicas por alteraciones en la hemostasia secundaria son poco frecuentes en la EvW, excepto en el tipo 3, en donde el factor VIII se encuentra muy reducido y los pacientes pueden tener hematomas y hemartrosis, semejante a los pacientes con hemofilia. La hemorragia después del parto es rara en los pacientes con EvW tipo 1, en el cual los niveles del FVIII/FvW son casi normales y generalmente se encuentran normales al final del embarazo. En pocos casos, los niveles del FVIII/ FvW no son normales durante el embarazo y estas mujeres requieren tratamiento profiláctico con desmopresina o concentrados de FVIII/FvW antes del parto. Las pacientes con EvW tipo 2A, 2B y 3 usualmente requieren tratamiento con terapia de reemplazo post-parto. Cuadro 1 Datos clínicos más frecuentes en los enfermos con EvW. Datos Clínicos Frecuencia de Presentación Epistaxis 60% Hemorragia transvaginal 50% Hemorragia Post-extrac50% ción dental Equimosis 40% Gingivorragias 35% Hemorragia Post-parto 20% Hemorragia 10% Gastrointestinal Hematuria 5% * 5% Hematomas * † Hemartrosis 3% /40% ‡ *Estos defectos ocurren más frecuentemente en el tipo 3. o en la EvW tipo Normandy. †se refiere al tipo 1 y algunos subtipos 2. ‡se presentan en tres estudios de pacientes con EvW tipo 3. DIAGNÓSTICO. Los pacientes con EvW manifiestan síntomas hemorrágicos que son típicos de defectos de hemostasia primaria. La enfermedad debe sospecharse en cualquier paciente con historia de hemorragia mucocutánea (epistaxis, metrorragias, gingivorragias, etc.) y postoperatoria, especialmente sí la historia familiar sugiere un patrón de herencia autosómica. Los pacientes con EvW tipo 3 presentan hemorragias que semejan la hemofilia: hemartrosis, hemorragias musculares, etc. (defectos de hemostasia secundaria). La interpretación de los valores de laboratorio del FvW es frecuentemente difícil, dado que el diagnóstico se establece con la imagen global de todas las pruebas de hemostasia. Por regla general, no hay un valor de corte aceptado en donde el paciente pueda Revista de Hematología Vol. 7, No. 1, 2006 24 S Quintana-González, C Martínez-Murillo. ser clasificado como EvW en forma definitiva. Existen además variaciones importantes de los niveles del FvW plasmático en el mismo paciente, variables como el ejercicio, el tabaquismo, enfermedad subyacente, fármacos (ejemplo los anticonceptivos orales) y el embarazo pueden modificar los niveles del FvW; el grupo sanguíneo ABO y otros antígenos fuera del sistema ABO como el Lewis. Debido a la variabilidad biológica de la EvW, el diagnóstico resulta difícil y únicamente logra establecerse después de varias determinaciones de las pruebas de hemostasia. Por lo tanto, con la variabilidad del FvW un solo valor normal no excluye la EvW en el paciente sintomático. Al igual, valores anormales deben confirmarse y repetir las pruebas posteriormente (14). Pruebas de Escrutinio. En el cuadro 2 se describen las pruebas de laboratorio empleadas para los pacientes con sospecha de EvW. El cuadro 3 señala la nomenclatura del complejo FVIII/FvW de acuerdo a las recomendaciones de la Sociedad Internacional de Hemostasia y Tr o m b o s i s ( I n t e r n a t i o n a l S o c i e t y o f Thrombosis and Hemostasis). En las pruebas de escrutinio la cuenta de plaquetas (CP) es usualmente normal, la trombocitopenia leve puede ocurrir en pacientes con tipo 2B. El (TH) usualmente esta prolongado, pero puede estar normal en pacientes con formas leves de la enfermedad como ocurre en el tipo 1. El tiempo de protrombina (TP) es normal y el tiempo de tromboplastina parcial activado (TTPa) puede estar prolongado de acuerdo a la concentración del FVIII. El FvW: Antigénico (FvW:Ag) y el cofactor de ristocetina (FvW:RiCof) son las pruebas Cuadro 2 Pruebas de laboratorio de la EvW. Pruebas de Escrutinio Pruebas para establecer el tipo de EvW Tiempo de hemorragia Agregación plaquetaria inducida por ristocetina (RIPA) TTPa Pruebas de unión al FvW (colágena y FVIII) FvW:RiCof Multímeros del FvW FvW:Ag Pruebas de FvW plaquetario FVIII:C Análisis de DNA Analizador de la función plaquetaria (PFA) Grupo sanguíneo ABO Cuadro 3 Nomenclatura del Complejo del Factor VIII/FvW propuesta por la International Society of Trombosis and Hemostasis. Factor VIII Proteína Antígeno Función Factor de von Willebrand Proteína Madura Antígeno Actividad Cofactor de Ristocetina Capacidad de Unión a la Colágena Capacidad de Unión al Factor Revista de Hematología Vol. 7, No. 1, 2006 VIII VIII:Ag VIII:C FvW FvW:Ag FvW:RCo FvW:CB VIII FvW:FVIIIB 25 Enfermedad de von Willebrand. Cuadro 4 Hallazgos de laboratorio en los tipos de la EvW. Tipo FVIII FvW:Ag FvW:Rcof RIPA Patrón Multimérico (plasma) 1 2A ↓ ↓ ↓ ↓ ↓ ↓↓ ↓ o normal ↓ 2B 2M 2N 3 ↓ o normal ↓ o normal ↓↓ ↓↓ Normal o ↓ ↓ Normal No detectado ↓↓ ↓↓ Normal No detectado ↑ ↓ o normal Normal ↓↓ Todos los tamaños presentes Ausencia de multímeros de tamaño intermedio y grandes Ausencia de grandes multímeros Todos los tamaños presentes Todos los tamaños presentes Ausencia total del FvW básicas para la EvW. Estudios adicionales como la agregación plaquetaria inducida por Ristocetina (RIPA) y el estudio de los multímeros permiten caracterizar a la EvW para un tratamiento apropiado (cuadro 4). De acuerdo a la caracterización del la EvW, tenemos las siguientes variedades: Tipo 1: Es la forma más común (70% de los casos) que se caracteriza por una disminución cuantitativa del FvW el cual es funcionalmente normal y representa un grupo muy heterogéneo de enfermedades. La mayoría de los tipos I no se logra explicar su defecto molecular. Clásicamente, el tipo I se hereda en forma autosómica dominante, pero existen algunas excepciones (18). La EvW tipo 1 se caracteriza por hemorragias leves a moderadas, TH normal o discretamente prolongado y niveles bajos de FvW:Ag, FvW:RiCof y FVIII, con multímeros presentes. Se tienen muchas dificultades para establecer los criterios diagnósticos estrictos en esta enfermedad. Un diagnóstico definitivo requiere niveles bajos del FvW en más de una ocasión (usando grupos sanguíneos ajustados al rango normal), historia de hemorragia e historia familiar positiva. Sin uno de los dos últimos criterios el diagnóstico debe considerarse como “probable” (19). Los valores bajos del FvW:Ag y FvW:RiCof son difíciles de evaluar, porque entre otros factores los niveles dependen del grupo ABO y el nivel de FvW:Ag esta disminuído aproximadamente en un 25% en personas con grupo sanguíneo “0” comparado con los otros grupos. En estos casos los pacientes compatibles con el tipo 1 son considerados cuando los niveles de FvW:Ag y FvW:RiCof se encuentran 2DS más abajo y ajustarlo de acuerdo al grupo sanguíneo. Tipo 2: Se refiere a deficiencias cualitativas del factor de von Willebrand. No existen datos sobre la incidencia correcta de esta enfermedad, sin embargo, se estima que de todos los tipos de EvW del 20-30% pertenecen al tipo 2. El tipo 2 es muy heterogéneo e incluye a 4 subtipos; 2A, 2B, 2M y 2N. 2A.- Se hereda en forma autosómica dominante. Las mutaciones se presentan en el dominio A2 que interfiere con el ensamblaje y el transporte intracelular de los grandes multímeros. Estos pacientes son identificados por niveles bajos o n o r m a l e s d e l F v W: A g y m a r c a d a m e n t e disminuídos los niveles de FvW:RiCof, con un patrón multimérico anormal, caracterizado por pérdida de los multímeros de alto peso molecular y un aumento en la intensidad de los multímeros d e b a j o p e s o m o l e c u l a r. E l s i t i o d e multimerización se localiza actualmente en los dominios D3-A1. El mecanismo detallado de las mutaciones A2 permanece sin explicación. Otras mutaciones localizadas en el dominio A2 se asocian con una elevada sensibilización de los multímeros a la proteólisis en la circulación. Otros pacientes presentan un tipo recesivo de Revista de Hematología Vol. 7, No. 1, 2006 26 S Quintana-González, C Martínez-Murillo. la enfermedad, con mutaciones en el dominio D2, que son compatibles con el papel propuesto del propéptido en la unión del puente de disulfuro, el cual es necesario para el proceso de multimerización (20). 2B. – Se caracteriza por un aumento de la afinidad del FvW por la GPIb de las plaquetas. Se detecta por la agregación plaquetaria a bajas concentraciones de ristocetina. Al igual que otros subtipos de la EvW, también es muy heterogénea en los niveles de FvW:Ag. El patrón multimérico se reporta con deficiencia de los multímeros de alto peso molecular y algunas veces trombocitopenia. Las mutaciones están localizadas en el dominio A1, la mayoría en la región N-terminal del asa de unión del puente disulfuro. Se hereda en forma autosómica dominante. 2M (Multímero).- La unión a plaquetas se encuentra afectada, pero el patrón multimérico es normal. Las mutaciones que se observan en este subtipo están localizadas en la región del exón 28 igual que en el tipo 2B, las mutaciones en este subtipo inactivan el sitio de unión para la unión a plaquetas o colágena. Los resultados de laboratorio son similares al subtipo 2A, pero el patrón multimérico las diferencia 2N (Normandy).- En este subtipo existe una disminución de la afinidad por el factor VIII, todas las mutaciones se localizan en la región N-terminal de la subunidad madura la cual contiene el sitio de unión del FVIII, en el dominio D’, aunque algunos casos son encontrados en el dominio D3. La enfermedad se hereda en forma recesiva. La función plaquetaria se encuentra normal, los niveles de FvW:Ag y FvW:RiCof son normales, la estructura multimérica es normal, pero los niveles de FVIII se encuentran disminuídos. La hemorragia en estos pacientes es causada principalmente por la disminución del FVIII:C y debe de diferenciarse de la hemofilia clásica leve. Revista de Hematología Vol. 7, No. 1, 2006 Tipo 3: La EvW tipo 3 es la variedad que originalmente informó en 1926 Erick von Willebrand y se define como la ausencia de FvW:Ag circulante, niveles disminuidos de FVIII:C (15%), se hereda en forma autosómica recesiva y es la forma más severa de la EvW. La prevalencia se estima en 1:1,000 000 de sujetos. Las hemorragias son caracterizadas no sólo por hemorragia mucocutánea sino también por hemartrosis y hematomas, como las que se observan en pacientes con hemofilia. Las mutaciones se han encontrado en el exón 18. Algunos casos del Tipo 3 resultan de deleciones completas o parciales del gene del FvW. Estos pacientes tienen predisposición para desarrollar aloanticuerpos (5-8%) por la presencia de deleciones, por lo tanto, es importante evaluar el riesgo del desarrollo de inhibidores (21). En México, se llevo a cabo un estudio con la finalidad de confirmar el diagnóstico y clasificar a pacientes con sospecha de EvW mediante el análisis del patrón multimérico. Estudiaron un total de 30 pacientes de los cuales 19 tuvieron tipo 1, 8 del tipo 2 y 3 la variedad tipo 3 (22). Muchas de las mutaciones, las cuales causan diferentes formas de EvW, se han identificado y correlacionan sus efectos sobre la estructura y f u n c i ó n d e l F v W. P o r o t r o l a d o , o t r a s enfermedades pueden estar relacionadas a defectos cuantitativos o cualitativos en el FvW, como la EvW adquirida (23) y la púrpura trombocitopénica trombótica recurrente (24). El FvW se ha asociado también con la trombosis arterial. Además es un marcador plasmático de la activación endotelial en algunas enfermedades vasculares crónicas, como las angiopatías en los pacientes con diabetes mellitus (25). TRATAMIENTO. El objetivo del tratamiento en la EvW es corregir los defectos de la hemostasia. Corregir las anormalidades en la hemostasia primaria 27 Enfermedad de von Willebrand. (adhesión y agregación plaquetaria) y los defectos de la hemostasia secundaria. El tratamiento debe cohibir la hemorragia o prevenirla en caso de un procedimiento quirúrgico. A diferencia de la hemofilia, la profilaxis regularmente no se utiliza en los pacientes con EvW, porque usualmente las hemorragias son menos severas. Sin embargo, en los pacientes con EvW tipo 3 tienen hemorragias más graves y presentan hemartrosis recurrentes, lo que ocasiona, al igual que la hemofilia artropatías. Por lo tanto, puede estar indicado en estos pacientes la profilaxis. La elección del tratamiento depende del subtipo de la EvW y la naturaleza de la diátesis hemorrágica (cuadro 5). A pesar de la alta prevalencia de la EvW, existen pocos estudios bien controlados sobre la duración e intensidad del tratamiento. Los niveles de FVIII deben tener un nivel hemostático adecuado de 30 UI/dL y el objetivo principal es corregir los defectos de la hemostasia primaria. La corrección del tiempo de sangrado y el incremento de los niveles de FvW:RiCof a 50 UI/dL son los parámetros más importantes. En el caso de la EvW tipo 3, en la cual el comportamiento es semejante a la hemofilia y tienen hemorragia por defectos de hemostasia secundaria, los niveles del FVIII debe estar entre 30-50 UI/dL, dependiendo del sitio de la hemorragia. Hay dos tratamientos de elección en la EvW: la desmopresina (DDAVP) y la terapia transfusional con productos sanguíneos. Entre los tratamientos adyuvantes están los inhibidores de la fibrinolisis, las preparaciones de estrógenos-progestágenos orales y las fibrinas adhesivas. Desmopresina (1-Deamino -8-D-Arginina vasopresina). La desmopresina es un derivado sintético de la hormona antidiurética, originalmente descubierto para el tratamiento de diabetes insípida. La desmopresina (DDAVP) es un agonista selectivo para el receptor V2 (V2R). Es probable que la desmopresina actúe sobre una célula intermedia que libera una hormona liberadora del FvW, la cual más tarde actúa sobre la célula endotelial. En pacientes con hemofilia leve y en algunos pacientes con enfermedad de von Willebrand, la desmopresina incrementa de manera transitoria los niveles plasmáticos del Factor VIII y FvW de los cuerpos de Weibel-Palade en las células endoteliales. También libera el Factor de plasminógeno tisular (t-PA) e interleucina-8 (IL8) (cuadro 6). Las ventajas de utilizar desmopresina es su costo relativamente bajo, con ilimitada disponibilidad y al ser un medicamento sintético no transmite enfermedades infecciosas. La desmopresina es administrada en niños y adultos a dosis de 0.3 microgramos/Kg de peso, en 20- Cuadro 5 Medidas terapéuticas en la EvW. Tipo de EvW Tratamiento de Elección 1 Desmopresina (DDAVP) 2A, 2M Concentrado de FVIII-FvW 2B 2N 3 Concentrado de FVIII-FvW Concentrado de FVIII-FvW Concentrados de FVIII-FvW Transfusión de plaquetas Tratamiento Secundario Concentrado de FVIII-FvW Crioprecipitados Crioprecipitados Desmopresina (DDAVP)????? Crioprecipitados Desmopresina (DDAVP) Crioprecipitados Revista de Hematología Vol. 7, No. 1, 2006 28 S Quintana-González, C Martínez-Murillo. Cuadro 6 Efecto farmacológico de la desmopresina (DDAVP). Incremento del Factor VIII Incremento del Factor de von Willebrand (FvW) Acorta el Tiempo de Hemorragia Mejora la adhesión y agregación plaquetaria Incrementa la GpIb-IX (¿?) Incrementa t-PA Incrementa la IL-8 Efectos no conocidos 30 ml de solución fisiológica, en infusión continua, durante 30 minutos por vía intravenosa, en promedio aumentará el Factor VIII y el Factor de von Willebrand de 3-5 veces de las concentraciones basales de estos factores, en un lapso de 30-60 minutos. La desmopresina también puede administrarse por via subcutánea a la misma dosis que la intravenosa y por inhalación nasal (cuadro 7). La administración subcutánea o intranasal son convenientes para tratamiento profiláctico y tratamiento en casa. La administración oral no ha sido evaluada para uso en pacientes con EvW. Para emplear la desmopresina es muy importante realizar previamente la prueba a la desmopresina antes de administrarla para uso terapéutico debido a que hay pacientes que no responden al tratamiento. Una vez que el paciente se ha comprado la respuesta al tratamiento, esta respuesta generalmente será consistente. La prueba terapéutica a la desmopresina se debe administrar a la misma dosis anteriormente señalada. Se obtiene un valor basal de los niveles de Factor VIII y FvW:RiCof o la prueba de unión a la c. Una vez que el paciente se ha comprado la respuesta al tratamiento, esta respuesta generalmente será consistente. Se administra la desmopresina y 3060 minutos después se determinan los factores, para conocer los niveles máximos de los factores y a las 4 horas para obtener la vida media. En caso de que los niveles de Factor VIII y FvW se encuentren Revista de Hematología Vol. 7, No. 1, 2006 entre el 10-20% de la actividad son suficientes para cubrir hemorragias leves a moderadas. Sin embargo, se requieren niveles de 30-50% de actividad, para extracciones dentales, pero no para cirugía mayor. Es muy importante evaluar los niveles de estos factores después de la administración de la desmopresina, para identificar en que tipo de hemorragia puede ser utilizada. La desmopresina puede emplearse cada 12-24 horas, si es necesario. Cuadro 7 Dosis y vías de administración de la desmopresina. Dosis: 0.3 microgramos/Kg peso/dosis en infusión continua durante 20-30 minutos IV o sc 300 microgramos vía Intranasal en adultos 150 microgramos vía intranasal en niños Niveles de Factor: 1.5 veces incrementa los niveles de factor VIII 3-5 veces incrementa los niveles de factor de von Willebrand Niveles máximos: 30-60 minutos vía IV 90-120 minutos vía intranasal o subcutánea Vida Media: 5-8 horas para factor VIII 8-10 horas para FvW 29 Enfermedad de von Willebrand. No se recomienda administrarla por más de tres días, por la liberación del t-PA. La respuesta a la desmopresina varía dependiendo del tipo de EvW y no en todos los pacientes con EvW esta indicado (cuadro 6). Los pacientes con EvW tipo 1, en la cual el factor de von Willebrand es funcionalmente normal, tienen mejor respuesta que los pacientes con EvW tipo 2. No se recomienda en el tipo 2A porque habrá un incremento disfuncional del FvW y no es efectivo para hemostasia primaria, aunque existen excepciones. La desmopresina está contraindicada en el tipo 2B, porque produce trombocitopenia después de la administración del medicamento. Los pacientes con el tipo 2M tienen poca respuesta a la desmopresina, pero en la práctica debe de hacerse la prueba terapéutica para decidir el tratamiento. En la EvW tipo 2N, existe aumento de los niveles del Factor VIII, pero la respuesta es a corto tiempo por el defecto en la unión con el FvW, por lo que no se recomienda como primera opción terapéutica. Los pacientes con EvW tipo 3 no tienen respuesta a la desmopresina, porque carecen de sitios de almacenamiento del FvW. Los efectos adversos de la desmopresina son secundarios a la vasodilatación cutánea, como la cefalea, enrojecimiento facial y tinitus; por lo general son leves. La presencia de hiponatremia por retención hídrica, causada por el efecto antidiurético de la desmopresina, es raro pero puede presentarse con la ingesta abundante de líquidos, por lo que se recomienda, evitar la ingesta excesiva de líquidos. El mayor problema de retención hídrica e hiponatremia con presencia de crisis convulsivas se observa en niños pequeños por lo que se debe de evitar el medicamento en niños menores de dos años de edad. No debe administrarse en pacientes con enfermedad arterial coronaria, porque al liberarse los grandes multímeros del FvW puede aumentar la agregación plaquetaria y puede causar infarto agudo del miocardio. Concentrados de factor VIII. Los pacientes con EvW que no pueden ser tratados con DDAVP requieren la sustitución de ambos factores: factor VIII y FvW. No todos los concentrados de Factor VIII contienen FvW. Entre los concentrados de Factor VIII que contienen FvW se encuentra el Humate-P, el cual se ha evaluado ampliamente en estudios clínicos. Contiene grandes cantidades de FvW, el concentrado está inactivado viralmente por medio de pasteurización. Otro concentrado de factor VIII que contiene grandes cantidades de FvW se encuentra el Alfanate, el cual es inactivado viralmente con solvente/detergente y altas temperaturas. El Inmunate también contiene factor VIII/FvW, derivado del plasma humano, es un concentrado de alta pureza con doble inactivación viral (tratamiento inicial con polisorbato 80 seguido de calentamiento con vapor durante 10 horas a 60ºC), el cual se ha demostrado su eficacia en el tratamiento de los pacientes con EvW26. Existen otros factores de factor VIII con FvW que aunque no existen grandes estudios clínicos, son efectivos clínicamente, entre ellos se encuentran el Octanate y Fandhi. Los concentrados monoclonales no contienen FvW al igual que el Factor VIII recombinante, los cuales no deben de emplearse en los pacientes con EvW, porque carecen del FvW. La dosis de los concentrados de factor VIII/ FvW depende del sitio de hemorragia. Se recomiendan en caso de hemorragias leves de 15 a 20 UI/Kg. Dependiendo de la severidad de la hemorragia se puede administrar cada 12 horas. En caso de extracción dental se recomienda una dosis de 20-30 UI/Kg. En cirugía de 40-50 UI/Kg diariamente; en cirugía mayor de 5-10 días de tratamiento y en cirugía menor cada tercer día usualmente, 2-4 días. Crioprecipitados. Los crioprecipitados contienen factor VIII, factor de von Willebrand, fibrinógeno, factor XIII y fibronectina. Los crioprecipitados son útiles en los pacientes con EvW porque contienen la glucoproteína multimérica del FvW. Sin embargo, en la actualidad no se recomiendan como primera Revista de Hematología Vol. 7, No. 1, 2006 30 S Quintana-González, C Martínez-Murillo. elección de tratamiento, porque no se inactivan viralmente y pueden transmitir infecciones. Las complicaciones del uso del crioprecipitado son los mismos riesgos de transmisión de enfermedades que el plasma fresco congelado. Pacientes que reciben grandes cantidades de crioprecipitados para hemofilia A y por largos periodos de tiempo (ejemplo procedimientos quirúrgicos) han tenido paradójicamente hemorragia a pesar de adecuados niveles de FVIII:C y niveles adecuados de FvW. Esta hemorragia está relacionada a altos niveles de fibrinógeno, que producen altas cantidades de productos de degradación de fibrina y alargamiento del TM. Antifibrinolíticos. Estos productos inhiben la activación del plasminógeno y la actividad de la plasmina, por lo tanto, previenen la lisis del coágulo. Los pacientes con enfermedad de von Willebrand frecuentemente presentan sangrado transvaginal y epistaxis, esto se debe a la gran actividad fibrinolítica que se presenta en estas mucosas. Los pacientes que se someten a extracciones dentales tienen gran actividad fibrinolítica local, que puede incrementar la presencia de hemorragia en estos pacientes. Los antifibrinolíticos se pueden administrar en forma sistémica o local, entre ellos se encuentran al ácido aminocaproico (Amicar) el cual se indica a dosis de 50-60 mg/Kg c/6 h, el ácido tranexámico (10-15 mg/Kg/8 h), puede administrarse VO, IV o tópica. En caso de hemorragias leves pueden usarse solos o coadyuvantes al tratamiento con concentrados de factor VIII/FvW o desmopresina. Están contraindicados en pacientes con hematuria ya que al no lisar el coágulo éste puede obstruir el tracto urinario. Estrógenos/progestágenos. La presencia de menorragias es frecuente en los pacientes con EvW, lo que causa anemia por deficiencia en hierro. En las pacientes con EvW tipo 3, la administración de estrógenos/progestágenos reduce hasta en el 88% la pérdida de sangre. El mecanismo de acción de estos medicamentos es Revista de Hematología Vol. 7, No. 1, 2006 hacer menos susceptible al endometrio de sangrar. Tratamiento en el embarazo. En mujeres con FvW tipo I, los niveles de factor VIII y FvW aumentan espontáneamente durante el embarazo y los niveles de estos factores son normales al termino del embarazo (27). El nivel del Factor VIII es el mejor predictor de hemorragia durante y después del embarazo. Por lo tanto, se recomienda medirlo al término del embarazo y dos semanas después, cuando los niveles del factor VIII disminuyen rápidamente y puede ocurrir hemorragia (28). El riesgo de hemorragia después del parto vaginal o cesárea es mínimo cuando los niveles de factor VIII se encuentran entre 30-40% de los niveles normales (28). En las pacientes con EvW tipo 3, no es necesario monitorear los niveles de factor VIII, porque estos no cambian durante el embarazo y la administración diaria de concentrados de Factor VIII/FvW deben ser administrados para evitar hemorragia (29). La dosis que se recomienda es 40 UI/Kg/día antes y de 3-4 días post-parto y tener niveles de factor VIII >50% del nivel normal (29). Tratamiento de pacientes con aloanticuerpos antifactor de von Willebrand. Pacientes con EvW tipo 3 pueden presentar aloanticuerpos contra el FvW después de múltiples transfusiones que contienen FvW (30). Estos anticuerpos se presentan con una frecuencia del 1015% de estos enfermos (31). La infusión de los concentrados de FvW son inefectivos y pueden causar anafilaxia post-transfusión debido a la formación de complejos inmunes circulantes. Los concentrados que contienen FvW están contraindicados después de esta complicación porque la presencia del FvW pone en riesgo la vida del paciente por ocasionar reacciones anafilácticas, por la activación del complemento y por la formación de los complejos inmunes (32). Para control de las hemorragias en los pacientes con aloanticuerpos, puede usarse concentrados de factor VIII recombinante, que no tienen el FvW. Sin embargo, la vida media de este factor transfundido es 31 Enfermedad de von Willebrand. muy corta (1-2 h) porque carece del FvW y, por lo tanto, se degrada rapadamente el FVIII transfundido. La recomendación es administrar el tratamiento en infusión continua, a grandes dosis, con el objetivo de mantener niveles hemostáticos del Factor VIII y controlar los fenómenos hemorrágicos (33). Existen estudios del uso del factor VIIa recombinante en estos pacientes y la dosis recomendada es de 90 microgramos/Kg cada 2 h o 20 microgramos/Kg cada hora, en infusión continua (34). Enfermedad de von Willebrand adquirida (EvWa). La primera descripción de la forma adquirida de la EvW fue en 1968 (35). La EvWa es una enfermedad hemorrágica poco común, con hallazgos clínicos y de laboratorio similares a la forma hereditaria de la enfermedad. Las primeras descripciones de pacientes con EvWa eran causadas por padecimientos inmunológicos (36). En los siguientes años, diferentes enfermedades clínicas produjeron disminución en los niveles del FvW que provocaban enfermedad hemorrágica. En los pacientes con EvWa el FvW es sintetizado en cantidades normales o sintetizarse en mayor cantidad y liberado normalmente a la circulación sanguínea. Los mecanismos propuestos en el desarrollo de la EvWa son la presencia de autoanticuerpos que forman complejos inmunes circulantes, lo que provoca la destrucción del FvW circulante. Otro mecanismo es la adsorción del FvW sobre las células neoplásicas u otras superficies celulares, aumento en la degradación proteolítica del FvW (cuadro 8). Las enfermedades frecuentemente asociadas con la EvWa son las enfermedades linfoproliferativas, autoinmunes o neoplasias. El tratamiento principal de los pacientes con EvWa es erradicar la causa subyacente. Otras opciones de tratamiento son la administración de DDAVP, concentrados de factor, globulina inmune intravenosa (37). CONCLUSIONES. Han existido avances importantes en el diagnostico y tratamiento de los pacientes con EvW. En México es necesario contar con laboratorios de referencia para completar y diagnosticar los subtipos de la EvW, ya que las opciones terapéuticas anteriormente descritas están disponibles. Actualmente, se encuentra en estudios la preparación recombinante del FvW. La interleucina-11, una citosina que se ha observado en animales y humanos, incrementar el factor VIII y el FvW, la cual puede ser una nueva forma de tratamiento en el futuro para los pacientes con EvW. Cuadro 8 Mecanismos patogenéticos en la EvWa. Autoanticuerpos específicos o no específicos que forman complejos inmunes circulantes e incrementan la destrucción del FvW: Enfermedades linfoproliferativas Enfermedades neoplásicas Enfermedades inmunológicas Adsorción del FvW sobre las clonas de las células neoplásicas u otras superficies celulares: Enfermedades linfoproliferativas Enfermedades neoplásicas Enfermedades mieloproliferativas Incremento del shear stress Incremento de la degradación proteolítica del FvW: Específico: Enfermedades mieloproliferativas Incremento del shear stress Uremia Ciprofloxacina No-específico (plasmina): Hiperfibrinolisis primaria Hiperfibrinolisis secundaria Lisis terapéutica Incremento Shear Stress: Alteraciones cardiacas congénitas Estenosis aórtica Endocarditis Malformación vascular: - Telangiectasia Hemorrágica Hereditaria - Síndrome de Kasabach-Merrit Ateroesclerosis severa β-talasemia Síntesis disminuida: Hipotiroidismo Desconocido: Acido valproico Enfermedad viral Trasplante de hígado Revista de Hematología Vol. 7, No. 1, 2006 32 S Quintana-González, C Martínez-Murillo. REFERENCIAS. 1.- Von Willebrand EA. Hereditär pseudohemofili. Finska Läkarsällskapetes Handl 1926; 68:87-112. Willebrand factor that mediates platelet adhesion on subendothelium is not collagen. J Clin Invest 1988; 82: 65-73. 2.- Nilsson IM, Blombäck M, Jorpes E, Blombäck B, Johansson SA. Von Willebrand’s disease and its correction with human plasma fraction 1-0. Acta Med Scand 1957; 159:179-88. 13.- Ruggeri ZM , Ware J. The structure and function of von Willebrand factor. Thromb Haemost 1992; 67: 594-599. 3.- Holmberg L, Nilsson IM. Genetic variants of von Willebrand’s disease. Br Med J 1972; 3:317-20. 4.- Hoyer LW, Shainoff JR,. Factor VIII-related protein circulates in normal human plasma as high molecular weight multimers. Blood 1980, 55;1056-59. 5.- Ruggeri ZM, Pareti FI, Manucci PM. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. N Engl J Med 1980, 302;1047-51. 6.- Sadler JE. A revised classification of von Willebrand disease. Thromb Haemost 1994; 71:520-25. 14.- Blömback M, Erenoth P, Andersson O, Anvret M. On laboratory problems in diagnosing mild von Willebrand disease. Am J Hematol 1992; 40:117-20. 15.- Cattaneo M, Federici AB, Lecchi A, Agati B, Lombardi R, Stabile F, et al. Evaluation of the PFA-100 system in the diagnosis and therapeutic monitoring of patients with Willebrand disease. Thromb Haemost 1999; 82: 35-39. 16.- Manucci PM, Lombardi R, Bader R. Heterogeneity of type 1 von Willebrand disease: evidence for a subgroup with abnormal von Willebrand factor. Blood 1985; 66:796-802. 7.- Rodeghiero F., Castaman C., Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood; 69:454-459. 1987. 17.- Gralnick HR, Rick ME, McKeown LP. Platelet von Willebrand factor. An important determinant of the bleeding time in type I von Willebrand disease. Blood 1986; 68:58-61. 8.- Nilsson IM., Borge L., Gunnarsson M., Kristoffersson AC. Factor VIII related activities in concentrates. Scand J Haematol; 33, Suppl 41: 157-172. 1984. 18.- Sadler JE, Matsushita T, Dong Z, Tuley EA, Westfield LA. Molecular mechanism and classification of von Willebrand disease. Thromb Haemost 1995; 74: 161-6. 9.- Mannucci PM, Bloom AL, Larrieu MJ, Nilsson IM, West RR. Artherosclerosis and von Willebrand factor. Prevalence of severe von Willebrand’s disease in western Europe and Israel. Br J Haematol 1984; 57:1639. 19.- Battle J, Torea J, Rendal E, Fernández MFL. The problem of diagnosing von Willebrand disease. J Intern Med 1997; 242: 121-8. 10.- Berliner SA, Seligson U., Zivelin A., Zwang E., Sofferman G. A relatively high frecuency of severe type 3 von Willebrand´s disease in Israel. Br J Haematol; 1:20-34. 1986. 20.- Eikenboom JCJ, Reitsma PH, Peerlinck KMJ, Briet E. Reccesive inheritance of von Willebrand’s disease type 1. Lancet 1993; 341: 982-6. 21.- Federici AB and Mannucci PM. Diagnosis and management of von Willebrand disease. Haemophilia 1999; 5: 28-37 11.- Jiménez C. Estudio epidemiológico de la enfermedad de von Willebrand en escolares en Costa Rica. Memorias de las XVI Jornadas Anuales de la Agrupación Mexicana para el Estudio de la Hematología. Puebla, México, 1995. 22.- Martínez-Murillo C, Viveros ME. Enfermedad de v o n Wi l l e b r a n d E n : M a n u a l d e H e m o s t a s i a y Trombosis. Martínez Murillo C, Quintana G, editores. México: Editorial Prado; 1996. 12.- De Groot PG, Ottenhof-Rovers M, van Mourik JA, Sixma JJ. Evidence that primary binding site of von 23.- Tefferi A, Nichols WL. Acquired von Willebrand disease: concise review of occurrence, diagnosis, Revista de Hematología Vol. 7, No. 1, 2006 33 Enfermedad de von Willebrand. pathogenesis, and treatment. Am J Med 1997; 103:53640. 24.- Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Laemmie B. Deficient activity of von Willebrand factor-cleaved protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood 1997; 89:3097-103. 25.- Stehouwer CD, Fischer HR, van Kuijk AW, Polak BC, Donker AJ. Endothelial dysfunction precedes development of microalbuminuria in IDDM. Diabetes 1995; 44: 561-4. 26.- Auerswald G, Eberspâcher B, Engl W, Güthner C, Koksch M, Kreuz W, et al. Successful treatment of patients with von Wllebrand disease using a highpurity double-virus inactivated factor VIII/von Willebrand concentrate (Inmunate®). Sem Thromb Hemost 2002; 28: 203-213. 27.- Noller KL, Bowie EJ, Kempers RD, Owen CA Jr. Von Willebrand's disease in pregnancy. Obstet Gynecol 1973; 41:865-72. III von Willebrand's disease and an inhibitor against von Willebrand factor. Haemostasis 1996;26:Suppl 1:150-4. 34.- Lak M, Peyvandi F, Mannucci PM. Clinical manifestations and complications of childbirth and replacement therapy in 385 Iranian patients with type 3 von Willebrand disease. Br J Haematol 2000; 111:1236-9. 35.- Simone JV, Cornet JA, Abildgaard CF. Acquired v o n Wi l l e b r a n d ’s S y n d r o m e i n s y s t e m i c l u p u s erythematosus. Blood 1968; 31: 806-811. 36.- Ingram GIC, Kingston PJ, Leslie J, Bowie EJW. Four cases of acquired von Willebrand’s syndrome. Br J Haematol 1971; 21: 189-199. 37.- Federici AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of acquired von Willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood 1998; 92:2707-11. 28.- Conti M, Mari D, Conti E, Muggiasca ML, Mannucci PM. Pregnancy in women with different types of von Willebrand disease. Obstet Gynecol 1986; 68:282-5. 29.- Mannucci PM. Drug Therapy: Treatment of von Willebrand’s Disease. N Engl J Med 2004; 351: 683-694 30.- Castaman G, Federici AB, Rodeghiero F, Mannucci PM. Von Willebrand’s disease in the year 2003: towards the complete identification of gene defects for correct diagnosis and treatment. Haematologica 2003; 88:94108. 31.- Mannucci PM, Meyer D, Ruggeri ZM, Koutts J, Ciavarella N, Lavergne JM. Precipitating antibodies in von Willebrand's disease. Nature 1976; 262:141-2. 32.- Bergamaschini L, Mannucci PM, Federici AB, Coppola R, Guzzoni S, Agostoni A. Posttransfusion anaphylactic reaction in a patient with severe von Wi l l e b r a n d d i s e a s e : r o l e o f c o m p l e m e n t a n d alloantibodies to von Willebrand factor. J Lab Clin Med 1995; 125:348-55. 33.- Ciavarella N, Schiavoni M, Valenzano E, Mangini F, Inchingolo F. Use of recombinant factor VIIa (NovoSeven) in the treatment of two patients with type Revista de Hematología Vol. 7, No. 1, 2006 35 Inhibidores adquiridos de los factores de la coagulación. Jaime García-Chávez, Lilia A. García-Stivalet. Unidad Médica de Alta Especialidad, Hospital de Especialidades del Centro Médico Nacional “La Raza”, Instituto Mexicano del Seguro Social, México, D.F., México. INTRODUCCIÓN. Los inhibidores adquiridos contra los factores de la coagulación son una manifestación más de el fenómeno de autoinmunidad, proceso causal de una morbilidad y mortalidad considerable. Generalmente se trata de anticuerpos dirigidos contra ciertos determinantes antigénicos de los factores de la coagulación específicos. Se han descrito tres tipos de anticuerpos: autoanticuerpos, aloanticuerpos y xenoanticuerpos. Los aloanticuerpos más comunes son aquellos que surgen en pacientes con deficiencias hereditarias de factores de la coagulación que son tratados con proteínas recombinantes o nativas. Son distintos a los autoanticuerpos que surgen de forma espontánea en personas sin coagulopatías hereditarias. Los xenoanticuerpos se desarrollan en pacientes que se exponen a factores de coagulación de origen animal (1). Los autoanticuerpos aparecen de forma frecuente en pacientes mayores y son atribuidos a una pérdida de la vigilancia inmunológica por anticuerpos anti-idiotípicos. Se pueden detectar títulos bajos de autoanticuerpos dirigidos contra el factor VIII en pacientes sanos. Generalmente están dirigidos contra el sitio activo del factos, aunque también se han descrito los inhibidores contra otros sitios, lo que condiciona un acortamiento notable de la vida media (2). Por otro lado, se pueden observar autoanticuerpos, llamados inhibidores espontáneos, responsables de producir manifestaciones hemorrágicas y alteraciones en las pruebas de coagulación. Los pacientes afectados pueden sufrir otras enfermedades de naturaleza autoinmune como el Lupus Eritematoso Sistémico (LES), aunque frecuentemente se trata de pacientes previamente sanos. También se puede asociar a otras enfermedades como cáncer, exposición a algunas drogas como penicilina (inhibidores contra FVIII y XII) estreptomicina o gentamicina (inhibidor contra factor V), isoniazida (inhibidor contra factor XIII). Algunos anticuerpos se encuentran en el posparto inmediato (p.e. inhibidores contra FVIII y FIX). Las pruebas de laboratorio muestran niveles bajos de uno o más factores de la coagulación. El diagnóstico se sospecha cuando al adicionar plasma del paciente a plasma normal, éste sufre un alargamiento de los tiempos de coagulación (1). Los a utoa ntic ue r pos que c o mp lic a n enfermedades como LES, enfermedades Solicitud de reimpresos: Dr. Jaime García-Chávez, Pestalozzi No. 635 int. 7, Col. Narvarte, C.P. 03020, México, D.F., México. Tel: 55 23 83 48 E-mail: jaimeg@prodigy.net.mx Revista de Hematología Vol. 7, No. 1, 2006 36 J García-Chávez, LA García-Stivalet. malignas o secundarias a fármacos, remiten con el tratamiento efectivo de la enfermedad de base. En cambio los que se catalogan como idiopáticos pueden desaparecer en forma espontánea mientras que otros persisten por años (1). De todos los inhibidores contra factores de la coagulación, aquellos que inactivan al factor VIII son los más frecuentes. INHIBIDORES CONTRA FACTOR VIII. Epidemiología. Los anticuerpos adquiridos contra el factor VIII son poco frecuentes, mientras la mayoría aparecen en pacientes mayores sin antecedentes de enfermedades concomitantes. También se asocian a LES, asma, embarazo y enfermedades neoplásicas. Se han descrito 40 casos de inhibidores contra factor VIII en asociación a cáncer. Los más comunes son: leucemia linfocítica crónica, aunque se han descrito asociación con tumores sólidos y otras enfermedades oncohematológicas (3). Inmunología. Los inhibidores contra el factor VIII son inmunoglobulinas del tipos IgG 1 e IgG 4 (4). La m a y o r í a so n de cadena ligera kap pa , y generalmente no se unen a complemento. Generalmente se dirigen contra el dominio A2 (en la cadena pesada), contra el dominio C 2 (en la cadena ligera), o ambos en la molécula del FVIII. Ensayos de inmunoprecipitación muestran que el 60% de los pacientes presentan múltiples anticuerpos contra FVIII. Sin embargo, los estudios de neutralización mostraron que se requiere generalmente de un solo fragmento para que se neutralice la actividad del inhibidor (1). Los anticuerpos que reaccionan contra el dominio A2 (más específicamente la región entre los aminoácidos 484 a 509), pueden interferir con la interacción entre esta subunidad del FVIII y el factor IXa. Se mostró una correlación inversa entre los niveles de Bethesda y la Revista de Hematología Vol. 7, No. 1, 2006 concentración de anticuerpo requerida para inhibir la estimulación dependiente de A2 del factor IXa (1). Los anticuerpos contra el dominio C 2 pueden interferir con la unión del FVIII a la fosfatidilserina, contra el factor de Von Willebrand o ambos (5). La actividad anticoagulante de este inhibidor puede resultar de la interferencia con la membrana de unión del FVIIIa, que se requiere para su integración al complejo Xasa en formación (6). Manifestaciones Clínicas. El síntoma de presentación más común es hemorragias a nivel de piel y músculos. Sin embargo, durante el curso de la enfermedad pueden ocurrir hemorragias en cualquier sitio de la economía. Muchos pacientes presentan hemorragias a nivel de mucosas o membranas; puede existir epistaxis o gingivorragias recurrentes. Otros pueden presentar melena o hematoquezia desarrollando síndrome anémico secundario. También se pueden manifestar como hematuria persistente, en especial asociado a infección de vías urinarias o enfermedad prostática. Se pueden desarrollar hematomas musculares en brazos y piernas secundarios a traumatismos leves con complicaciones como compresión de nervios o compromiso de la irrigación arterial a las extremidades. Las hemartrosis son menos comunes, aunque pueden ocurrir. En pacientes en el posparto inmediato la manifestación más importante es la hemorrgia transvaginal persistente (cuadro 1). La hemorragia retroperitoneal puede ser masiva y producir la muerte por choque hipovolémico. La hemorragia intracraneal es poco frecuente, pero con consecuencias devastadores. Las hemorragias mayores ocurren generalmente en más del 80% de los casos, con una mortalidad aproximada de 20% (1). En resumen se debe sospechar en un inhibidor de FVIII en pacientes mayores con hemorragias espontáneas o hemorragia después de un trauma menor. 37 Inhibidores adquiridos de la coagulación. observa en pacientes con hemofilia, enfermedad de Von Willebrand o deficiencia de factores de contacto. Sin embargo, se distingue de estos realizando correcciones con plasma normal. En los pacientes con deficiencias, la adición de plasma normal corrige el TTPa, pero cuando se adiciona plasma normal a un plasma que contiene inhibidor, el TTPa se prolonga. Esto se debe realizar inmediatamente después de la mezcla y después de la incubación a 37°C por una hora. Este procedimiento permite que los inhibidores débiles que requieren de más tiempo para la incativación del FVIII sean reconocidos (1). Las no corrección con plasma normal proporcionan evidencia de que existen inhibidores contra el FVIII, pero se dispone de pruebas específicas para cada factor y también se puede cuantifivcar la potencia de mismo por diferentes técnicas como la de Bethseda y sucedáneas (figura 1) (1). Cuadro 1 Sitios frecuentes de hemorragia en pacientes con inhibidores contra factor VIII. Sitio: % Músculo Piel Vías urinarias Tubo digestivo Articulaciones Heridas quirúrgicas Orofaringe e hipofaringe Otors 44 13 9 9 6 6 4 9 Diagnóstico. El dato pivote en el diagnóstico de inhibidores contra FVIII es un tiempo de tromboplastina parcial activada (TTPa) prolongado, sin alteraciones sobre el tiempo de protrombina (TP). Este patrón también se ÿ TTPa prolongado + TP normal + Cuadro clínico compatible ÿÿÿÿ ÿ ÿÿÿ ÿÿÿÿÿ ÿÿ ÿÿÿ Figura 1.- Algoritmo para el diagnóstico de inhibidores contra FVIII. Revista de Hematología Vol. 7, No. 1, 2006 38 J García-Chávez, LA García-Stivalet. Tratamiento (figura 2). Tratamiento durante el evento hemorrágico. Se cuenta con diferentes recursos terapéuticos, cada vez más efectivos, si bien hace apenas unos 5 años el pronóstico era muy distinto. El Factor VIII porcino, fue por mucho tiempo el tratamiento de elección, pero ahora ya ha salido del (7). Los efectos adversos eran poco frecuentes, los más comunes eran mialgias y dolor lumbar, así como menos frecuentemente anafilaxia. La incidencia reportada de reacciones adversas es de 10 por 1000 infusiones en pacientes que reciben menos de 100UI/kg y 82 por 1000 infusiones en pacientes que reciben dosis más altas (1). Otro efecto secundario reportado con el FVIII porcino es trombocitopenia, la cual puede deberse a agregación plaquetaria ex vivo (1). Cuando el tiempo lo permite se deben esperar los resultados de la prueba de Bethesda, basando el tratamiento en los niveles de inhibidor. Si encontramos un nivel menor a 5 U.B., se debe administrar FVIII humano iniciando con bolos de 100 UI/kg seguido de una infusión continua de 10 UI/Kg/h hasta el control de la hemorragia o bien que sea claro que el tratamiento no está siendo efectivo. Se deben determinar concentraciones plasmáticas del factor a las 4 a 6 h de iniciado el tratamiento para asegurar niveles plasmáticos mayores a 0.25 UI/ml. En caso de persistir con hemorragia o con niveles bajos de FVIII, esto sugiere que la potencia del inhibidor es mayor a la estimada por la prueba de Bethesda o que el FVIII en el concentrado es muy susceptible a la inactivación por el inhibidor. El FVIII que se encuentra en concentrados que tienen niveles altos de factor de von Willebrand (p.e. concentrados de pureza intermedia) pueden encontrarse protegidos de la inactivación por algunos inhibidores, que tienen una mayor especificidad por el dominio C 2 del factor VIII (1). Por lo que en estos casos el tratamiento debe ser cambiado a este tipo de concentrados. Revista de Hematología Vol. 7, No. 1, 2006 En los casos en los que la hemorragia no se controla con los concentrados de FVIII existen dos alternativas: el factor VII activado humano y recombinante (rFVIIa) y el concentrado de complejos activados, que contienen protrombina, factor VII, IX y X. El rFVIIa ofrece varias ventajas potenciales. Es efectivo iniciando la formación del coagulo cuando se une al factor tisular, este complejo subsecuentemente activa al FIX y X. Sin embargo, a dosis terapéuticas también se puede unir directamente con la fosfatidilserina plaquetaria de forma independiente del factor tisular (8). Bajo estas circunstancias se puede generar trombina en la superficie de las plaquetas activadas. Su efectividad hemostática es independiente del FVIII y por lo tanto no se afecta por los inhibidores del mismo. Otra ventaja es que no es un producto de la sangre humana, por lo que el riesgo de transmisión de agentes infecciosos es bajo. La experiencia clínica con el rFVIIa se analizó por Glazer y col. (8) en el cual se incluyeron 1270 episodios de hemorragia en 240 pacientes, 18 de estos pacientes con inhibidores. Se observaron respuestas efectivas en 74-100% de los pacientes dependiendo del tipo de hemorragia, cuando ésta era considerada como crítica la efectividad fue de 91%. Los efectos adversos fueron raros e incluyeron hipertensión, rash cutáneo, fiebre, cefalea y epistaxis. La dosis recomendada fue de 90 µg/kg, tomando en cuenta que la vida media del rFVIIa es de solamente 2.9 hrs, se debe repetir la dosis cada 2 a 3 h por 1 a 2 días o bien hasta la mejoría clínica. También se puede utilizar infusión continua del rFVIIa a una dosis de 16.5-20 µg/kg/h siendo segura y efectiva (9). Los concentrados del complejo protrombinasa estan aprobados por la FDA para el tratamiento de pacientes con inhibidores del FVIII (1). Se pueden encontrar 2 productos: Autoplex y FEIBA. Los procoagulantes activados presentes en Autoplex incluyen los factores VIIa, IXa, Xa, XIa, y trombina. 39 Inhibidores adquiridos de la coagulación. Mientras que FEIBA también contiene factor VIIa. La dosis utilizada de Autoplex es de 50U/ Kg y de FEIBA 50-75 U/Kg, las dosis se repiten cada 8 a 12 h. Con Autoplex se logran respuestas buenas o excelentes en 87% en pacientes con hemofilia, siendo su uso en pacientes con autoanticuerpos anecdótico pero en general positivo (1). En un estudio retrospectivo con FEIBA en el que se tratan 60 pacientes, 6 de ellos con autoanticuerpos contra el FVIII, se encontraron respuestas favorables en 81%, presentándose efectos adversos en 5 pacientes (1). No se deben administrar agentes antifibrinolíticos en pacientes que reciben concentrado de complejo protrombinasa. Estos concentrados son preparados utilizando sangre humana, por lo que potencialmente pueden transmitir agentes infecciosos. Inmunomodulación. La administración de globulina hiperinmune (IVIg) ha inducido disminución de los niveles de inhibidor en algunos pacientes. En un ensayo multicéntrico, 2 de 19 pacientes presentaron una disminución de los niveles de inhibidor de forma rápida, y 4 más tuvieron una respuesta gradual en el transcurso de algunos meses (10). La dosis utilizada fue de 1mg/kg/día por 2 días o 0.4g/ Kg día por 5 días. Adicionalmente los pacientes recibieron prednisolona 1mg/Kg/día por 2 semanas con posterior disminución de la dosis. Cua tr o pa c ie nte s pr e se nta r on r es p u e s ta completa, con niveles indetectables de inhibidores al día 21 de tratamiento. Los efectos adversos son poco frecuentes e incluyen mareos y prurito más frecuentemente, aunque ha habido reportes de insuficiencia renal aguda. El efecto de la IVIg se atribuye a la presencia de anticuerpos anti-idiotipo en el ÿ ÿÿ µ ÿÿ ÿ ÿÿ ÿ ÿ ÿÿÿÿÿ ÿÿÿÿÿ ÿ ÿ ÿÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿ ÿÿÿ ÿÿÿ ÿ ÿÿÿÿÿ ÿÿ ÿÿ ÿ ÿÿ Figura 2.- Algoritmo para el tratamiento de pacientes con inhibidores contra el FVIII. Revista de Hematología Vol. 7, No. 1, 2006 40 J García-Chávez, LA García-Stivalet. "pool" de inmunoglobulinas utilizado para prepararla. Estos anticuerpos se dirigen contra los autoanticuerpos del paciente. Otras opciones incluyen plasmaféresis o inmunoadsorción. La plasmaféresis por sí sola no es efectiva debido a que se encuentran grandes reservorios extravasculares de autoanticuerpos, por lo que se deben cambiar por lo menos 4 litros de plasma para tener un impacto sobre los niveles de anticuerpos. La inmunoabsorción implica el uso de columnas extracorpóreas que contienen proteínas adsorbentes de inmunoglobulinas unidas a sefarosa, con reducciones de 76% de los niveles de inhibidor posterior a sesiones de 4 h. Tratamiento a largo plazo. En aproximadamente una tercera parte de los pacientes, generalmente en aquellos sin enfermedades asociadas, los anticuerpos desaparecen de forma espontánea (1). Sin embargo, la pérdida del anticuerpo puede llevar meses a años, durante los cuales el paciente se encuentra en riesgo de presentar hemorragias graves. Cuando la presencia del anticuerpo se asocia a una enfermedad subyacente, el tratamiento de la misma puede llevar a la desaparición del anticuerpo. Sin embargo en la mayoría de los pacientes las enfermedades subyacentes no pueden ser erradicadas (p.e. cáncer). Bajo estas circunstancias se debe intentar eliminar el inhibidor. Cuando se el diagnóstico de inhibidor se confirma se debe administrar prednisona 1mg/ Kg/día. Una respuesta satisfactoria es la disminución del nivel de inhibidor en 3 semanas a menos del 50% de su valor inicial. Si esto ocurre se debe continuar el tratamiento hasta que el inhibidor desaparezca y se encuentren niveles normales de FVIII. Esto se observa en una tercera parte de los pacientes (1). Los pacientes que responden tienen un nivel menor de inhibidor que los pacientes que no responden (3 UB contra 50 UB respectivamente) y niveles más Revista de Hematología Vol. 7, No. 1, 2006 altos de FVIII (9% contra 1%). En los 2/3 de pacientes que tienen persistencia del inhibidor se encuentra varias opciones: 1. Suspender prednisona e iniciar ciclofosfamida a 2mg/Kg/día por 3 a 6 semanas. 2. Continuar prednisona y adicionar ciclofosfamida. 3. Iniciar ciclosporina con o sin otro agente a dosis de 5mg/Kg/día para lograr niveles plasmáticos de 150-350ng/ml 4. Uso de quimioterapia de combinación con prednisona (p.e. Ciclofosfamida + vincristina). Los inhibidores que aparecen en el posparto, casi todos los casos se resuelven en los 30 meses posteriores, asociándose más frecuentemente con primeros embarazos y provocando hemorragias transvaginales u de otro tipo inmediatamente posparto, con tendencia a reaparecer en los siguientes embarazos no ha sido efectivo. El tratamiento con esteroides y ciclofosfamida, auque este tratamiento no ha sido del todo efectivo. Otros tratamientos. Rituximab: El anticuerpo monoclonal anti CD-20, rituximab, elimina de forma rápida la mayoría de las células B circulantes. El éxito en el tratamiento de los linfomas con rituximab lo ha vuelto atractivo como candidato para el tratamiento de las enfermedades benignas que involucran los linfocitos B. El concepto central de esto es la remoción de la fuente celular de los anticuerpos patológicos, se ha utilizado este tipo de tratamiento en enfermedades como PTI con algunos éxitos. Se han realizado algunos ensayos clínicos utilizando rituximab como tratamiento para la hemofilia adquirida, mostrando efectividad como tratamiento de primera línea, así como en pacientes refractarios a otros tratamientos inmunosupresores. Se encontró depleción de los linfocitos B circulantes, observándose mejores respuestas en pacientes 41 Inhibidores adquiridos de la coagulación. con títulos de Bethesda bajos (<100BU/ml) con desaparición completa de la actividad del inhibidor en 3 a 12 semanas. Las recaídas se asociaron a un nivel de inhibidor más altos, aunque con el reinicio del tratamiento se lograron nuevas remisiones completas (11). Por lo que se puede concluir con la información con la que se cuenta hasta el momento que el uso de rituximab como t r a t a m i e n to es efectivo, requirie ndo su combinación con otros agentes en pacientes con niveles altos de inhibidor (11). INHIBIDORES CONTRA FACTOR VON WILLEBRAND. La deficiencia de factor de von Willebrand (FvW) puede ser congénita o adquirida. Las causas de deficiencia adquirida incluyen defectos en la síntesis o liberación, absorción de la proteína por superficies celulares (neoplasias) degradación mecánica o proteolítica. Los casos inmunológicos incluyen aloanticuerpos y a u t o a n t i c u e r p o s c o n t r a e l F v W. L o s autoanticuerpos pueden ser idiomáticos o bien la primera manifestación de una enfermedad autoinmune como LES o linfomas. El anticuerpo generalmente se une a sitios no funcionales de l a p r o t e í n a d e l F v W, p r e d i s p o n i e n d o a hemorragias cuando el complejo Ac-FvW es eliminado de forma rápida de la circulación. El diagnóstico debe ser sospechado en pacientes con episodios de hemorragia de aparición súbita, especialmente en sitios mucocutáneos como tracto gastrointestinal o genitourinario. Los laboratorios de rutina muestran un TTPa prolongado. Estudios más específicos mostraran una agregometría a ristocetina alterada y disminución de loa concentración de los multímeros de alto peso molecular del FvW. El tratamiento en estos casos con desmopresina frecuentemente controla la hemorragia. En los casos refractarios, se deben administrar concentrados de FvW. En pacientes con enfermedades asociadas, p.e. MGUS la inmunoglobulina hiperinmune provee una mejoría mas sostenida. Otro tratamiento utilizado es el recambio plasmático con buenos resultados, sin embargo el tratamiento a largo plazo se logra solamente con el tratamiento de la enfermedad subyacente con desaparición de los anticuerpos. INHIBIDORES CONTRA FACTOR V. Este tipo de inhibidores son raros, ocurren de forma espontánea en pacientes de la tercera edad, muy frecuentemente en pacientes que han sido sometidos recientemente a procedimientos quirúrgicos (13). Otras asociaciones encontradas es la exposición a aminoglucósidos, transfusiones y enfermedades malignas. Los inhibidores contra el factor V frecuentemente son anticuerpos IgG policlonales (1). El diagnóstico se sospecha con base en un TTPa y TP prolongados los cuales no corrigen con la adición de plasma normal. Se pueden observar niveles muy bajos de factor V en pacientes con anticoagulante lúpico, con inhibidores específicos contra el factor V. Las manifestaciones hemorrágicas, en pacientes con inhibidores contra factor V, pueden ser desde triviales hasta poner en riesgo la vida. En caso de que se requiera tratamiento suele ser necesario el apoyo transfusional con plaquetas, ya que el factor V plaquetario se protege de los anticuerpos presentes en la c i r c u l a c i ó n ( 1 4) . O t r a s m e d i d a s , c o m o inmunoglobulina intravenosa, plasmaféresis, esteroides y agentes inmunosupresores, pueden resultar benéficos. INHIBIDORES CONTRA PROTROMBINA Y TROMBINA. Los anticuerpos dirigidos contra protrombina ocurren en pacientes con anticoagulante lúpico y algunos pueden forma complejos con protrombina que acelera su aclaración de la circulación. Las manifestaciones hemorrágicas son raras y la presencia de hemorragia ocurre solamente en los Revista de Hematología Vol. 7, No. 1, 2006 42 J García-Chávez, LA García-Stivalet. casos en los que se desarrolle hipoprotrombinemia grave. Por otro lado, los anticuerpos dirigidos contra la trombina se asocian a hemorragias. El anticuerpo descrito es un IgG que se une a la t ro m b i n a p e r o no a la protrombina . La hemorragia generalmente es de tipo mucocutánea y en sitios de punción. INHIBIDORES CONTRA FACTOR XIII. Posterior a la activación por la trombina y calcio, el factor XIII estabiliza la molécula de fibrina. Los anticuerpos interfieren con la actividad del factor XIII. Las manifestaciones hemorrágicas ocurren debido a que el coagulo recién formado es frágil, sufriendo lisis temprana. El diagnóstico debe sospecharse en aquellos pacientes con manifestaciones hemorrágicas y tiempos de coagulación normales. El diagnóstico se debe confirmar con la observación de que los coágulos formados por la recalcificación del plasma del paciente son solubles en 5M de urea o ácido monocloroacético al 1%. La mayoría de los pacientes son de edad avanzada, siendo afectados tanto hombres como mujeres. Los anticuerpos formados eran IgG los cuales evitan la estabilización de la fibrina por el factor XIII. El tratamiento es con concentrados del factor XIII lo cual neutraliza al anticuerpo y normaliza la solubilidad del coagulo. 4.- Fulcher CA, Mahoney S, Zimmerman TS. FVIII inhibitor IgG subclass and FVIII polypeptide specificity determined by inmunobloting. Blood 1987; 69:1475-80. 5.- Pratt KP, Shen BW, Takeshima K et al Structure of tha C2 domain of human factor VIII al 1.5 A resolution. Nature 1999; 402: 439-42. 6.- Scandella D, Gilbert GE, Shima M, et al. Some factor VIII inhibitor antibodies recognize common epitope corresponding to C2 domain amino acids 2248 through 23212 which overlap a phosholipid –binding site. Blood 1995; 86:1811-9. 7.- Morrison AE, Ludlam CA, Kessler C. Use of porcine factor VIII in the treatment of individuals with acquired hemophilia. Blood 1993; 81:1513-20. 8.- Monroe DM, Hoffman M, Oliver JA, et al. Platelet activity of high dose factor VIIa is independent of tissue factor. Br J Hematol 1997; 99:542-7. 9.- Santagostino E, Gringeri A, Morfini M, et al. Continuous infusion of recombinant activated factor VIIa for high titer inhibitor patients. Blood 2000; 96:267ª 10.- Schwartz RS, Gabriel DAAledort LM, et al. A prospective study of treatment of acquired (autoimmune) factor VIII inhibitor with high dose intravenous gama globulin. Blood 1995; 86:797-804. 11.- Stasi R, Brunetti M, Stipa E, et al. Blood 2004; 103:4424-8. 12.- Wiestner A, Cho H, Asch A, et al. Blood 2002; 100:3426-8. REFERENCIAS. 1.- Eckman MH, Erban JK, Singh SK, Kao GSJ. Screening for the risk for bleeding or thrombosis. Ann Intern Med 2003; 138: W 15-24. 2.- Giles J, Arnout J, Vermylen J, et al. Anti-factor VIII antibodies of hemophiliac individuals are frequently directed towards nonfunctional determinants and do not exibit isotypic restricction. Blood 1993; 82:245261. 3.- Sallh S, Wan JY. Inhibition against factor VIII in patients with cancer. Analysis for 41 patients. Cancer 2001; 91:1607-74. Revista de Hematología Vol. 7, No. 1, 2006 13.- Feinstein DI, Green D, Federici AB, et al. Diagnosis and managment of patients with spontaneously acquired inhibitors of coagulation. Hematology 1999:192-208. 14.- Schleinitz N, Veit V, Chowguet D, Seux V, Arnoux D, Mokat D, et al. Adquired factor V inhibitor: etiology, bleeding risk and therapeutic management with regar to three cases. Rev Med Interne 2001; 22:1119-23. 43 Coagulación intravascular diseminada. Abraham Majluf-Cruz. Depto. de Hematología, Hospital Regional No. 222 "Gabriel Mancera", México, D.F., México. INTRODUCCIÓN. La coagulación intravascular diseminada (CID) también se denomina coagulopatía por consumo. Es un síndrome que siempre es secundario a una enfermedad primaria (1). La CID representa una de las principales causas de morbilidad y mortalidad en el mundo. Es la primera causa de muerte en las unidades de terapia intensiva en los Estados Unidos de Norteamérica y la undécima causa de muerte (2). Cada año aparecen más de 750,000 casos nuevos de sepsis grave (>500 muertes/día por sepsis grave). La CID es compleja e impredecible, asociada con una mortalidad entre 28% y 50%. La población afectada es muy heterogénea y su característica fundamental es la imposibilidad de predecir su curso clínico (3). El incremento en la incidencia se origina del aumento de la población y su edad; el incremento en la frecuencia de infecciones nosocomiales; la resistencia bacteriana a los antibióticos cada vez más frecuente, a pesar de que éstos son cada vez más potentes; la elevación del número de pacientes inmunocomprometidos secundario a los nuevos tratamientos para el cáncer o las enfermedades autoinmunes, además de las nuevas causas de inmunosupresión natural; y el aumento en el número de cirugías cada vez más extensas y agresivas. La CID se caracteriza por activación del siste ma de c oa gula c ión, que r e s u lta e n generación intravascular de fibrina que puede ocluir los vasos sanguíneos pequeños y medianos y comprometer el suplemento sanguíneo a los órganos y que puede contribuir a la aparición de falla orgánica múltiple. El consumo de plaquetas y proteínas del sistema de coagulación, inducen hemorragia de intensidad variable. La definición de CID es: síndrome adquirido, caracterizado por la activación intravascular del sistema de coagulación hasta la formación intravascular de fibrina proceso que puede acompañarse de hiperfibrinolisis secundaria o de hipofibrinolisis (4). ENTIDADES CLÍNICAS ASOCIADAS A LA CID. La CID es secundaria a otra enfermedad primaria. La CID aumenta el riesgo de muerte más allá del de la enfermedad primaria. Además, el control de la causa primaria no necesariamente alivia la CID. Las infecciones bacterianas son las enfermedades más frecuentemente asociadas Solicitud de reimpresos: Dr. Abraham Majluf-Cruz, Jaina 41, Col. Letrán Valle, C.P. 03650, México, D.F., México. Tel: 55 39 13 58 E-mail: amajlufc@prodigy.net.mx Revista de Hematología Vol. 7, No. 1, 2006 44 A Majluf-Cruz. Cuadro 1 Condiciones clínicas asociadas a la CID. Condición clínica Causa Sepsis o infección grave Potencialmente, cualquier micro-organismo Traumatismos Los que cursan con lesión tisular grave, traumatismos de la cabeza y embolismo graso Destrucción de órganos Pancreatitis grave Neoplasias Sólidas, hematológicas (como la leucemia aguda promielocítica) Complicaciones obstétricas Placenta abrupta y embolia de líquido amniótico Alteraciones vasculares Hemangiomas gigantes o aneurismas gigantes. Falla hepática De cualquier causa: viral, tóxicos, medicamentos Reacciones tóxicas o inmunológicas graves Reacción transfusional aguda, rechazo de trasplantes, uso de drogas ilícitas y mordedura de serpientes además de las infecciones sistémicas por cualquier otro microorganismo. La CID es resultado de la gravedad de la infección con una respuesta inflamatoria generalizada, caracterizada por la liberación citocinas (5). Los traumatismos graves son otra condición asociada a CID, ya que liberan material tisular a la circulación (grasa o fosfolípidos) y generan hemólisis y daño endotelial, en especial, los traumatismos a nivel de la cabeza. Las neoplasias sólidas o hematológicas cursan con CID por mecanismos poco entendidos, aunque la mayoría de los estudios implican al factor tisular. En la leucemia promielocítica aguda se encuentra una forma distinta de CID, la cual se caracteriza por un estado hiperfibrinolítico grave, aunado a un sistema de coagulación ampliamente activado (6) y aunque predomina la hemorragia, en la autopsia se encuentra trombosis diseminada en Revista de Hematología Vol. 7, No. 1, 2006 muchos pacientes. La CID aparece en entidades obstétricas agresivas: placenta abrupta y embolia de líquido amniótico (7). Éste activa la coagulación in vitro. El grado de separación placentaria correlaciona con la gravedad de la CID por la fuga de material similar a la tromboplastina placentario. Las alteraciones vasculares, como los aneurismas o los hemangiomas gigantes, pueden generar una activación local de la coagulación. Los factores hemostáticos activados puede causar CID, pero es más común la depleción sistémica de los mismos, así como de las plaquetas, lo cual puede resultar en una condición clínica que es indistinguible de la CID. FISIOPATOLOGÍA DE LA CID. Existen múltiples mecanismos causantes de la CID y ahora se tiene mayor claridad acerca 45 Coagulación intravascular diseminada. Figura 1.- Pérdida del balance del sistema de coagulación en la sepsis y la CID. Este imbalance se debe principalmente a la activación de la hemostasia y de la fibrinolisis y a la caída simultánea de los mecanismos anticoagulantes naturales. IaTP-1: inhibidor del activador tisular del plasminógeno tipo 1. de su etiología (figura 1). Directamente, existen mecanismos que p e r m i t e n no sólo la generación sino la persistencia en la generación de la trombina, enzima clave en el proceso de la CID. Deben considerarse los siguientes mecanismos: Generación descontrolada de trombina. In vivo, el control del sistema hemostático es crucial al balancear las actividades procoagulante y anticoagulante. La formación de fibrina es controlada por el sistema de la proteína C y por la antitrombina. Este balance hemostático que coordina la generación de trombina se pierde en la CID. La generación de trombina se detecta entre tres y cinco horas luego de la infusión de microorganismos o endotoxina y el complejo factor tisular/factor VIIa tiene un papel central. No existen cambios en la activación del sistema de contacto (8) y su inhibición no previene la activación del sistema hemostático. La eliminación del complejo factor tisular/factor VIIa con anticuerpos monoclonales para el factor tisular o el FVIIa inhibe la generación de trombina y previene la aparición de la CID y la mortalidad (9). Diseminación y mantenimiento de la generación de trombina. Aunque el factor tisular juega un papel fundamental en el inicio de la generación de trombina, otros procesos diseminan la c o a g u l a c i ó n i n t r a v a s c u l a r. L o s b r o t e s secundarios de trombina se generan en la vía intrínseca llevando al consumo y depleción de proteínas anticoagulantes (proteína C y antitrombina). La exposición a fosfolípidos negativos facilita más el ensamblaje y propagación Revista de Hematología Vol. 7, No. 1, 2006 46 A Majluf-Cruz. de factores hemostáticos (10). Estos mecanismos forman una respuesta expansiva en el tiempo y en el espacio, característica de la CID. Supresión de los anticoagulantes naturales y deterioro de la fibrinolisis. Contribuyen a la formación y mantenimiento de la generación de fibrina. La concentración plasmática de la antitrombina disminuye en el paciente séptico por consumo dependiente de la misma generación de trombina, de degradación por elastasa de los neutrófilos y por disminución de su síntesis hepática. La caída de la antitrombina se asocia con un aumento en la mortalidad. También ocurre una disminución del sistema de la proteína C por disminución en la síntesis de trombomodulina (11). El factor tisular es normalmente inhibido por el inhibidor de la vía del factor tisular, cuya administración inhibe la generación de trombina inducida por endotoxina y disminuye potentemente la mortalidad. Por otra parte, en el momento de la activación máxima de la hemostasia el sistema fibrinolítico está muy deteriorado. La bacteremia y la endotoxemia aumentan la actividad fibrinolítica, probablemente por liberación de activadores endoteliales del plasminógeno. A esta respuesta profibrinolítica sigue una supresión de la actividad fibrinolítica, con aumento del inhibidor del activador tisular del plasminógeno tipo 1 (12). Activación inflamatoria. Las alteraciones hemostáticas y fibrinolíticas dependen de citocinas inflamatorias: FNT-α, la interleucina 1 (IL-1) e interleucina 6 (IL-6). El principal mediador de la activación de la coagulación en la CID es la IL-6 (13). El FNTα influencia la activación por su efecto sobre la IL-6 y es el factor más importante en la disregulación de los anticoagulantes naturales y la fibrinolisis. Las citocinas anti-inflamatorias tales como la IL-10 modulan la activación de la coagulación. Una vez activados, el sistema inflamatorio y el de la coagulación amplifican más la respuesta. Mientras que las citocinas y otros Revista de Hematología Vol. 7, No. 1, 2006 mediadores pro-inflamatorios inducen activación de la coagulación, la trombina y otras proteasas interactúan con receptores localizados sobre células y generan más activación celular con el aumento inflamatorio consecuente. Si el proceso se generaliza escapa de la vigilancia local diseñada para controlar el proceso y el balance fisiológico se rompe, lo que mantiene el círculo inflamación:coagulación. Activación y disfunción endotelial. La respuesta normal del endotelio regula la coagulación y la inflamación (14). Su disfunción y falla puede llevar y quedar marcada por el desarrollo de la CID. La hipercoagulabilidad y el predominio de la hemorragia o trombosis dependen de factores genéticos y otros relacionados con el huésped. DIAGNÓSTICO DE LA CID. No existe una prueba única para hacer el diagnóstico certero. Sin embargo, la combinación de algunas pruebas de laboratorio ayuda a establecer con mayor certeza este diagnóstico (15). El diagnóstico debe basarse en la cuenta plaquetaria, evaluación global del coagulograma (tiempos de tromboplastina parcial activada y de protrombina), cuantificación de la concentración de antitrombina y de uno o dos factores hemostáticos y una prueba para detectar productos de degradación de la fibrina. Se recomienda utilizar los criterios diagnósticos que se muestran en el cuadro 2. Cuadro 2 Puntaje para el diagnóstico de la CID. Variable Cuenta plaquetaria Metabolito de la fibrina elevado Tiempo de protrombina prolongado Fibrinógeno 0 >100 no Puntaje 1 <100 moderado 2 <50 grave < 3 seg 3 a 6 seg > 6 seg > 1 g/L < 1 g/L 47 Coagulación intravascular diseminada. Debe calcularse en todo paciente en riesgo de tener este problema y que tiene alguna de las patologías mencionadas en el cuadro 1. Si el puntaje es >5, se hace el diagnóstico de CID establecida y el puntaje debe repetirse diariamente. Si es <5, es puntaje es sugestivo de CID pero no lo afirma. Debe entonces repetirse cada 1 a 2 días. Realizar las pruebas en serie es más útil que aisladamente. La reducción plaquetaria o la caída significativa serial son datos sensibles aunque no específicos de CID. El alargamiento del coagulograma puede reflejar consumo y depleción de factores, lo que se objetiviza mejor al cuantificar algún factor. La exactitud de las pruebas coagulométricas en un tiempo es limitada. Cuantificar la antitrombina evalúa el consumo del inhibidor más importante de la trombina, aunque es caro y requiere tecnología especializada. La cuantificación del fibrinógeno puede sugerir, pero no es muy útil para el diagnóstico, ya que actúa como un reactante de fase aguda, por lo que su concentración plasmática puede no disminuir. El dímero D es útil para diferenciar la CID de procesos con plaquetas bajas y alargamiento de los tiempos de coagulación. El diagnóstico de CID puede hacerse combinando pruebas de laboratorio de rutina (cuadro 2) (15). Los resultados normales no excluyen una CID, sin embargo, pueden terminar en un acortamiento de los tiempos de coagulación y en un incremento del fibrinógeno. Por esto, se sugiere la identificación temprana de la CID, no sólo con resultados anormales, sino también identificando su tendencia. Se requieren pruebas más específicas que relacionen inflamación y CID que deben ser simples y rápidas. TRATAMIENTO DE LA CID. Existen múltiples controversias y estudios clínicos apropiados por la complejidad de la CID y sus consecuencias. A pesar de esto, la piedra angular del tratamiento es el manejo específico y vigoroso de la causa de la CID. En algunos casos, la CID resuelve en las horas siguientes a la resolución de la afección primaria. En otros casos, la CID puede estar presente por algunos días aún después de que se instituyó el tratamiento. Las medidas de soporte son muy necesarias: sustitución plasmática y plaquetaria, anticoagulantes e inhibidores fisiológicos de la hemostasia. Terapia con plasma y plaquetas. La disminución de las plaquetas como de la concentración de los factores aumenta el riesgo hemorrágico. La sustitución con plaquetas o plasma no se indica sólo por los resultados de laboratorio y deben emplearse sólo en enfermos con hemorragia activa, en los que requieren de un procedimiento invasivo y en los que existe un alto riesgo de hemorragia. Anticoagulantes. Al menos, la heparina inhibe parcialmente la activación del sistema de coagulación en la sepsis y en otras causas de CID aunque la evidencia proviene de estudios de series de casos no controlados y su efecto benéfico sobre criterios de respuesta duros nunca se ha demostrado (16). La seguridad de la heparina es cuestionable en pacientes con riesgo hemorrágico alto. El anticoagulante ideal para la CID debe estar dirigido contra el factor tisular. Recientemente, se desarrolló un inhibidor específico y potente del complejo factor tisular/factor VIIa/ factor Xa (rNAPc2) que se obtiene de nemátodos. Actualmente, se realizan estudios fase II y III para analizar su efecto en la CID. Concentrados de inhibidores naturales. Ya que la antitrombina es el inhibidor fisiológico más importante de la trombina, se inició su uso en humanos. En estudios clínicos en pacientes con sepsis y/o choque séptico, se demostró mejoría del puntaje y acortamiento de la CID y mejoría orgánica. La caída de la proteína C contribuye en la CID la cual se asocia con resultados fatales. Se ha intentado suplementar con proteína C o drotrecogina Revista de Hematología Vol. 7, No. 1, 2006 48 A Majluf-Cruz. encontrándose un efecto favorable en estudios animales y en humanos (17). Finalmente, en vista del papel central que tiene el factor tisular en el inicio de todo el proceso fisiopatológico de formación de trombina, puede especularse que la administración del inhibidor de la vía del factor tisular de tipo recombinante pudiera ser muy útil en estos casos. REFERENCIAS. 1 . - M a r d e r V J , M a r t i n S E , F r a n c i s C W, e t a l . Consumptive thrombohemorrhagic disorders. In: Colman RW, Hirsh J, Marder VJ, et al. Editors. Hemostasis and thrombosis: basic principles and clinical practice. Philadelphia: JB Lippincott; 1987. p. 975–1015. 2.- Sands KE, Bates DW, Lanken PN, Graman PS, Hibberd PL, Kahn KL, et al. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA 1997; 278:234-40. 3.- Angus D, Wax R. Epidemiology of sepsis: an update. 7Crit Care Med 2001; 29(Suppl 1):S109-S116. 4.- Müller-Berghaus G, Madlener K, Blombäck M, (Eds). DIC: pathogenesis, diagnosis and therapy of disseminated intravascular fibrin formation. Amsterdam: Elsevier Science Publishers BV; 1993. 5.- Levi M, van der Poll T, ten Cate H, et al. The cytokine-mediated imbalance between coagulant and anticoagulant mechanisms in sepsis and endotoxemia. Eur J Clin Invest 1997; 27:3–9. 6.- Falanga A, Consonni R, Marchetti M, et al. Cancer procoagulant and tissue factor are differently modulated by all-trans-retinoic acid in acute promyelocytic leukemia cells. Blood 1998; 92:143–51. 7.- Martin JN, Stedman CM. Imitators of preeclampsia and HELLP syndrome. Obstet Gynecol Clin North Am 1991; 18:181–98. 8.- van der Poll T, Buller HR, ten Cate H, et al. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med 1990; 322:1622–7. Revista de Hematología Vol. 7, No. 1, 2006 9.- De La Cadena RA, Majluf-Cruz A, Stadnicki A, Colman RW, Suffredini AF. Activation of the contact and fibrinolytic systems after intravenous administration of endotoxin to normal human volunteers: correlation with the cytokine profile. Immunopharmacology 1996; 33:231-7. 10.- Lobato-Mendizabal E, Majluf-Cruz A. Trombofilia, tromboembolia y el uso de las heparinas no fraccionadas y de bajo peso molecular. Rev Invest Clin 2000; 56:346-65. 11.- Conway EM, Rosenberg RD. Tumor necrosis factor suppresses transcription of the thrombomodulin gene in endothelial cells. Mol Cell Biol 1988; 8:5588–92. 12.- Biemond BJ, Levi M, ten Cate H, et al. Endotoxininduced activation and inhibition of the fibrinolytic system: Effects of various interventions in the cytokine and coagulation cascades in experimental endotoxemia in chimpanzees. Clin Sci 1995; 88:587–94. 13.- van der Poll T, Levi M, Hack CE, et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med 1994; 179:1253–9. 14.- Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003; 101:3765-77. 15.- Taylor FB, Toh CH, Hoots WK, Wada H, Levi M. Towards definition, clinical and laboratory criteria and a scoring system for disseminated intravascular coagulation. Thromb Haemost 2001; 86:1327-30. 16.- Corrigan JJ Jr. Heparin therapy in bacterial septicemia. J Pediatr 1977; 9:695–700. 17.- Majluf-Cruz A, Bergés-García A, García-Chávez J. Hemofilia. En: Góngora-Biachi RA, editor. Hematología. Actualización 2004. Mérida: Agrupación Mexicana para el Estudio de la Hematología, A.C.; 2004;51-54.