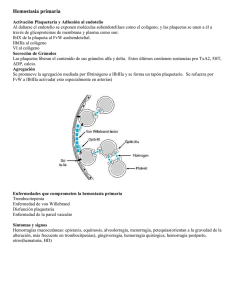
Hemostasia Primaria: Fisiología, Trastornos y Exploración Biológica
Anuncio

E – 1-1272 Hemostasia primaria A. Rauch, C. Paris Los trastornos de la hemostasia primaria reúnen un conjunto de anomalías plaquetarias, plasmáticas o vasculares, de origen constitucional o adquirido. La intensidad del fenotipo hemorrágico cutaneomucoso es extremadamente variable según la etiología causal y la gravedad del déficit. La exploración biológica inicial se basa en un número limitado de exámenes en busca de una trombocitopenia, una trombocitopatía o una enfermedad de Von Willebrand. Según el contexto, son necesarios otros exámenes, a veces muy especializados, de segunda línea para caracterizar una trombocitopatía o una trombocitopenia constitucional. El tiempo clínico es fundamental para orientar los estudios complementarios. © 2017 Elsevier Masson SAS. Todos los derechos reservados. Palabras clave: Hemostasia primaria; Trombocitopenia; Trombocitopatía; Enfermedad de Von Willebrand Plan activación plaquetaria es un proceso complejo, en el que están implicados receptores y vías de señalización múltiples y complementarios [1] . ■ Introducción 1 ■ Fisiología de la hemostasia primaria Principales actores de la hemostasia primaria Formación del trombo plaquetario 1 1 2 Principales actores de la hemostasia primaria ■ Semiología de los trastornos de la hemostasia primaria 3 Pared vascular ■ Exploración biológica de los trastornos de la hemostasia primaria Hemograma y estudio del frotis sanguíneo Lugar de las pruebas globales que exploran la hemostasia primaria Exploración de una trombocitopenia Exploración de una enfermedad de Von Willebrand Exploración de una trombocitopatía 3 3 3 4 5 5 ■ Etiologías de los trastornos de la hemostasia primaria Trombocitopenia Enfermedad de Von Willebrand Trombocitopatías Anomalías vasculares 6 6 9 10 11 ■ Conclusión 11 Introducción La hemostasia primaria es el proceso fisiológico que permite la formación del trombo plaquetario en respuesta a una lesión vascular. En la íntima, túnica en contacto con la luz vascular, se distinguen el endotelio, constituido por una monocapa de células endoteliales, y el subendotelio, formado por tejido conjuntivo y células musculares lisas. El endotelio en contacto con la sangre es tromborresistente, porque produce diferentes moléculas que inhiben la activación plaquetaria (prostaglandina I2, monóxido de nitrógeno), regula negativamente la coagulación (trombomodulina, inhibidor de la vía del factor tisular [TFPI]) o tiene una actividad profibrinolítica (activador tisular del plasminógeno [tPA]). El subendotelio, rico en moléculas adhesivas (colágeno, factor de Von Willebrand), es espontáneamente trombógeno. Factor de Von Willebrand El factor de Von Willebrand (FVW) es una glucoproteína sintetizada por las células endoteliales y los megacariocitos. Está presente en la sangre en una forma multimérica que soporta la adhesión y la agregación plaquetaria en la microcirculación en presencia de fuerzas de cizalladura elevadas [2] . También interviene en la coagulación estabilizando el factor VIII (FVIII) que, en ausencia de FVW, se elimina prematuramente de la circulación. Plaqueta Fisiología de la hemostasia primaria La iniciación, la amplificación y la estabilización de la activación plaquetaria se basan en la activación de receptores plaquetarios por moléculas adhesivas y agonistas solubles. La EMC - Tratado de medicina Volume 22 > n◦ 1 > marzo 2017 http://dx.doi.org/10.1016/S1636-5410(17)87866-1 Las plaquetas son fragmentos citoplasmáticos anucleados de 3 m de diámetro que proceden de la fragmentación citoplasmática de células hiperploides situadas en la médula ósea, los megacariocitos (MC). La maduración de los MC se produce siguiendo tres etapas sucesivas, asociadas a remodelaciones del citoesqueleto: la poliploidización del núcleo por un mecanismo de endomitosis, la formación de proplaquetas a partir de las 1 E – 1-1272 Hemostasia primaria Formación del trombo plaquetario membranas de demarcación y la migración de los MC hacia el nicho vascular, donde las plaquetas se liberan al flujo sanguíneo. La trombopoyetina (TPO) es el principal factor de crecimiento que estimula la megacariopoyesis. Interactúa con los receptores membranarios Mpl expresados por los progenitores MC y las plaquetas. La concentración de TPO se correlaciona inversamente con el nivel de expresión de Mpl por la masa MC/plaquetas. Numerosos factores de transcripción modulan los genes implicados en la megacariocitopoyesis en diferentes estadios de diferenciación (GATA-1, friend leukemia integration 1 [FLI-1], factor de transcripción relacionado con Runt-1 [RUNX-1], factor nuclear, factor nuclear eritroide 2 [NF-E2], etc.) [3] . La envoltura membranaria plaquetaria está constituida por una bicapa fosfolipídica de distribución asimétrica, con un predominio de fosfolípidos aniónicos en la lámina interna, exteriorizados durante la activación plaquetaria, que contienen receptores glucoproteicos esenciales para la fisiología plaquetaria. Un citoesqueleto submembranario mantiene la plaqueta en reposo en una forma discoide y permite la adquisición de una forma equinoide después de la activación plaquetaria. El citoesqueleto comporta dos redes de canales membranarios: el sistema canalicular abierto (SCA), constituido por invaginaciones profundas de la membrana plasmática, que facilita el despliegue plaquetario y la exocitosis rápida del contenido granular en caso de activación plaquetaria, y el sistema tubular denso, lugar de almacenamiento del calcio. Existen tres tipos de granulaciones citoplásmicas: los gránulos densos (ricos en calcio, trifosfato de adenosina [ATP], difosfato de adenosina [ADP], pirofosfatos, serotonina e histamina), los gránulos ␣ (ricos en factores de coagulación, factores de crecimiento y proteoglucanos) y los lisosomas (ricos en enzimas proteolíticas, como las catepsinas, las hidrolasas o las heparinasas). Durante la activación plaquetaria, se secretan los gránulos densos y ␣, y liberan su contenido. Moléculas adhesivas FVW a GPI GP1b a GPV GP1b a GP1b X GPI Colágeno GPVI X Lyn LAT a GP1b GPIb/IX/V En condiciones de flujo arterial, la captación y la adhesión inicial de las plaquetas en el subendotelio se efectúan principalmente por medio del complejo membranario glucoproteína (GP)Ib-IX-V, que tiene como ligando al FVW. En presencia de fuerzas de cizalladura elevadas, el FVW unido al colágeno subendotelial adopta una conformación que le permite unirse a la GPIb␣ plaquetaria [2] . Esta interacción permite una adhesión reversible de las plaquetas al subendotelio. La adherencia iniciada por la interacción GPIb␣-FVW se estabiliza después gracias a los receptores del colágeno (GPVI, ␣21). La acción de los receptores plaquetarios GPIb-IX-V y GPVI desencadena la activación plaquetaria a través de cascadas de señalizaciones intracelulares que convergen hacia la activación de la fosfolipasa C (FLC)␥2 y la producción de dos segundos mensajeros: el trifosfato de inositol (IP3) y el diacilglicerol (DAG) (Fig. 1). Estos dos mediadores activan respectivamente la movilización cálcica y la proteína-cinasa C (PCC) necesarias para la activación de la integrina ␣IIb3. En la transducción de la señal desencadenada por la unión del colágeno a la GPVI, expresada en la superficie de las plaquetas en forma de un complejo no covalente con la cadena ␥ común a los receptores de los fragmentos constantes de las inmunoglobulinas (FcR), intervienen cinasas de la familia Src (Fyn y Lyn), que fosforilan las dos tirosinas del motivo de inmunorreceptor activable por tirosina (ITAM) de la cadena ␥ del FcR. Los motivos ITAM fosforilados permiten la captación de otra tirosina-cinasa (Syk), que induce la formación de un complejo de señalización que conduce después a la activación de la fosfoinositida-3-cinasa (PI3K) y de la PLC␥2 [1] . La unión del FVW a la GPIb␣ en condiciones de flujo desencadena también una cascada de señalización que contribuye a la activación de la integrina ␣IIb3 igualmente por medio de la PI3K y de la PLC␥2 [1] . PI3K Kindlina-3 3§ 314- αIIbβ3 Fibrinógeno Syk Scr Akt Ta l PI3K Ca2+ a Rap1GTP PLCγ2 Akt β3 in CalDAG GEFI RASA3 AMPc Rap1GPD Rho-GEF FLCβ AC Pl3K ATP Gi G12/13 PAR1, PAR4, TP Gq P2Y12 PAR1, PAR4, TP, P2Y1 Agonistas solubles TXA2, trombina ADP Figura 1. Principales receptores implicados en la activación plaquetaria en respuesta a los agonistas solubles y a las moléculas de adhesión y que inducen un cambio de conformación de la integrina ␣IIb3 que permite su unión al fibrinógeno soluble (señalización dentro-fuera). GP: glucoproteína; FVW: factor de Von Willebrand; ADP: difosfato de adenosina; ATP: trifosfato de adenosina; AMPc: monofosfato de adenosina cíclico; TXA2: tromboxano A2; FLC: fosfolipasa C; PI3K: fosfatidilinositol 3-cinasa; AC: adenilato-ciclasa; PAR1, 4: receptor activado por proteasas 1, 4; CalDAG-GEF1: factor intercambiador de nucleótidos de guanina-1 regulado por diacilglicerol y calcio; RASA3: proteína activadora de GTPasa Ras-3. 2 EMC - Tratado de medicina Hemostasia primaria E – 1-1272 La activación plaquetaria se acompaña de una modificación de la morfología plaquetaria, indispensable para las reacciones de secreción y de agregación. La plaqueta pasa de discoide a esférica, con emisión de seudópodos y centralización de los orgánulos intracelulares. La centralización de los gránulos y después la fusión de sus membranas con la del SCA permite una liberación rápida del contenido granular. Las plaquetas activadas en contacto con la brecha vascular liberan localmente agonistas solubles como el ADP y el tromboxano A2 (TXA2), que amplifican la captación y la activación de las plaquetas cercanas, principalmente por medio de receptores de siete dominios transmembranarios acoplados a proteínas G (RAPG). Los principales RAPG implicados en la activación plaquetaria son los receptores de la trombina (receptor activado por proteasas [PAR]1 y PAR4), del ADP (P2Y1 y P2Y12) y del TXA2 (TP␣). La trombina, el TXA2 y el ADP inducen la activación de la FLC y la formación de los segundos mensajeros IP3 y DAG por medio de los receptores PAR1, PAR4, TP␣ y P2Y1 acoplados a proteínas Gq. P2Y12 y PAR1 también se acoplan a receptores Gi, que inducen una eliminación de la inhibición de la activación plaquetaria regulando negativamente diferentes inhibidores fisiológicos, como el monofosfato de adenosina cíclico (AMPc) y la molécula RASA3 [4] . El ADP y la trombina también desempeñan un papel esencial, a la vez en la iniciación (por medio de la vía Gq) y el mantenimiento de la activación plaquetaria (por medio de la vía Gi). Los receptores PAR1, PAR4 y TP␣ también se acoplan a receptores G12/13 implicados en la remodelación del citoesqueleto de actina-miosina a través de RhoA y sus efectores (Fig. 1). La integrina ␣IIb3, específica de las plaquetas, es un receptor esencial para la agregación plaquetaria. En la plaqueta en reposo, la ␣IIb3 está en un estado de baja afinidad, incapaz de unirse al fibrinógeno soluble. La activación plaquetaria por los agonistas fisiológicos induce una señalización dentro-fuera que da lugar a un aumento de la expresión membranaria y a un cambio de conformación de la ␣IIb3. Los receptores de la ␣IIb3 activados, agrupados en grupos en la superficie plaquetaria, adquieren la capacidad de fijarse al fibrinógeno y al FVW, lo cual permite la formación de puentes interplaquetarios. El FVW es el ligando preferido de la ␣IIb3 en presencia de fuerzas de cizalladura elevadas, condición reológica observada en la microcirculación [5] . La unión del fibrinógeno y el FVW a la ␣IIb3 también es el origen de una vía de señalización fuera-dentro (outside-in), que aumenta la estabilidad del agregado plaquetario y la retracción del coágulo. Las plaquetas activadas externalizan, en la lámina externa de su membrana plasmática, fosfolípidos aniónicos que constituyen una superficie procoagulante esencial para la captación de los complejos enzimáticos de la tenasa y la protrombinasa. La trombina formada amplifica la activación de las plaquetas en el seno del trombo por medio de los receptores PAR y permite la formación de un polímero de fibrina que estabiliza el trombo plaquetario. Semiología de los trastornos de la hemostasia primaria Debe buscarse un trastorno de la hemostasia primaria ante manifestaciones hemorrágicas espontáneas cutáneas (púrpura, equimosis) o mucosas (epistaxis, gingivorragias, ampollas endobucales, menorragias, etc.) o ante un síndrome hemorrágico posoperatorio de carácter inmediato. Esta expresión clínica difiere de las enfermedades de la coagulación que se manifiestan por hemorragias que afectan a los tejidos profundos (hematomas, hemartrosis) o que aparecen de manera retardada en el posoperatorio. Una púrpura corresponde a máculas eritematosas que resultan de la extravasación espontánea de los hematíes fuera de los capilares de la piel o las mucosas. La púrpura persiste con la vitropresión, lo cual la diferencia de los eritemas, los angiomas o las telangiectasias. Reviste diferentes aspectos: petequial, equimótica o, más raramente, víbices. La púrpura es patognomónica de un trastorno de la hemostasia primaria y plantea el problema de su etiología: anomalía cuantitativa o cualitativa de las plaquetas o enfermedad vascular. La púrpura trombocitopénica suele asociar petequias y equimosis. Las trombocitopatías y la enfermedad de Von WilleEMC - Tratado de medicina brand se manifiestan por equimosis y casi nunca por una púrpura petequial. La púrpura de origen vascular se caracteriza por una púrpura petequial, infiltrada, localizada en la zona declives y sin hemorragia mucosa asociada. La anamnesis debe precisar los antecedentes hemorrágicos personales y familiares, los antecedentes familiares de hemopatías, una posible consanguinidad familiar, la edad de inicio, el carácter espontáneo o provocado de los sangrados, los antecedentes quirúrgicos y transfusionales, los antecedentes de anemia y de suplementación con hierro, así como los tratamientos medicamentosos en curso. La exploración física debe orientarse también hacia la búsqueda de signos extrahemorrágicos a favor de una enfermedad plaquetaria sindrómica hereditaria (dismorfia, anomalías óseas, eccema, sordera, etc.), de un trastorno del tejido conjuntivo (hiperlaxitud cutaneoarticular, trastornos de la cicatrización, telangiectasias) o de una enfermedad subyacente (insuficiencia hepática, conectivitis, hemopatía). Estos elementos permiten orientar el diagnóstico etiológico en cuanto a la naturaleza del déficit y su origen constitucional o adquirido. Exploración biológica de los trastornos de la hemostasia primaria La exploración de primera línea se basa en un número limitado de exámenes. La solicitud de pruebas de coagulación es sistemática para descartar una coagulopatía. La exploración inicial puede evidenciar de entrada una trombocitopenia o una enfermedad de Von Willebrand, que requiere una exploración específica, u orientar hacia una trombocitopatía. Sin embargo, una exploración normal no permite descartar una trombocitopatía moderada. En caso de fuerte sospecha de trombocitopatía, es necesario realizar pruebas complementarias (Fig. 2). Hemograma y estudio del frotis sanguíneo Una trombocitopenia se define por un recuento plaquetario inferior a 150 g/l. Los tubos de hemograma contienen un anticoagulante seco, el ácido etilendiaminatetraacético (EDTA), que puede inducir in vitro la formación de cúmulos plaquetarios causantes de una seudotrombocitopenia. Las máquinas de recuento no tienen en cuenta las plaquetas de estos cúmulos, sólo cuentan las plaquetas libres. La medición automatizada del recuento de plaquetas y del volumen plaquetario medio no siempre es fiable, en particular en presencia de macroplaquetas, que pueden no haberse tenido en cuenta a causa de su tamaño. Por lo tanto, es indispensable un examen microscópico del frotis sanguíneo para confirmar o no el recuento plaquetario obtenido por la máquina y buscar un sesgo de análisis (aglutininas plaquetarias, macroplaquetas) o posibles anomalías que orienten hacia una etiología específica de trombocitopatía o trombocitopenia. Lugar de las pruebas globales que exploran la hemostasia primaria A pesar del interés diagnóstico limitado y de la ausencia de valor predictivo del riesgo hemorrágico, el tiempo de obturación plaquetaria (TOP) se utiliza con frecuencia para detectar un trastorno de la hemostasia primaria [6] . Comparado con el obsoleto tiempo de sangría in vivo, el TOP realizado in vitro tiene la ventaja de ser más reproducible y menos invasivo. Se realiza con una máquina, el Platelet Function Analyzer, Siemens (PFA-100), que mide la capacidad de las plaquetas de adherirse y agregarse, en presencia de fuerzas de cizalladura elevadas, a una membrana recubierta de colágeno y de un activador plaquetario (adrenalina o ADP). El TOP es sensible al recuento plaquetario, al hematocrito y a diferentes variables preanalíticas. La interpretación de un TOP debe tener en cuenta el hemograma, porque una trombocitopenia inferior a 100 g/l o un hematocrito inferior al 30% pueden inducir una prolongación del TOP. El TOP es un examen muy sensible para el 3 E – 1-1272 Hemostasia primaria Trastorno de la hemostasia primaria Exploración física Búsqueda de trombocitopatía Estudio: función plaquetaria por agregometría óptica, cuantificación de las GP membranarias por citometría Normal1 Anormal Anomalía vascular Trombocitopatía Ausencia Descartar una coagulopatía, TP, TPTA, fibrinógeno Anormal Coagulopatía Estudio de trombocitopenia, recuento de plaquetas Trombocitopenia confirmada en citrato Trombocitopenia Detección de enfermedad de Von Willebrand FVW:AG, FVW:RCo, FVIII:C ± TOP y RIPA Anormal Enfermedad de Von Willebrand Figura 2. Árbol de decisiones. Diagnóstico de un trastorno de la hemostasia primaria. GP: glucoproteína; TP: tiempo de protrombina; TPTA: tiempo parcial de tromboplastina activada; FVW:Ag: antígeno del factor de Von Willebrand; FVW:RCo: actividad de cofactor de la ristocetina; FVIII:C: factor VIII coagulante; TOP: tiempo de obturación plaquetaria; RIPA (agregación plaquetaria inducida por ristocetina): medición de la agregación de un plasma rico en plaquetas a diferentes concentraciones de ristocetina. 1. Completar según el contexto con el estudio de un marcador de secreción de los gránulos densos. diagnóstico de la enfermedad de Von Willebrand y de ciertas trombocitopatías graves, como la trombastenia de Glanzmann (TG), el síndrome de Bernard-Soulier (SBS) o la seudoenfermedad de Von Willebrand plaquetaria. Sin embargo, un TOP normal no permite descartar una trombocitopatía moderada, ni una variante 2N de la enfermedad de Von Willebrand [7] . Así pues, la realización sistemática de un TOP para detectar una trombocitopatía hereditaria carece de interés, debido a una sensibilidad y una especificidad insuficientes [8, 9] . “ Punto importante Limitaciones de las pruebas globales que exploran la hemostasia primaria • El tiempo de sangría y el TOP carecen de valor predictivo del riesgo de hemorragia. • Una anemia o una trombocitopenia pueden producir una prolongación del TOP. • Un TOP normal no permite descartar una trombocitopatía moderada. Exploración de una trombocitopenia Después de excluir una seudotrombocitopenia por EDTA, el diagnóstico etiológico se orienta por el contexto clínico y un número limitado de exámenes biológicos (Fig. 3). En ausencia de anomalías clínico-biológicas, se hace el diagnóstico por eliminación de púrpura trombocitopénica inmunológica (PTI). La PTI del niño es de aparición brusca y, al contrario que la PTI del adulto, su evolución suele ser favorable. Se realiza sistemáticamente un mielograma en caso de alteración del estado general, dolor óseo, síndrome tumoral, anomalías cuantitativas o cualitativas de las otras estirpes celulares del frotis sanguíneo. En caso 4 de médula pobre en el mielograma, puede ser necesaria una biopsia osteomedular. En ausencia de signos de alerta, el mielograma ya no se realiza sistemáticamente en el niño, excepto si se quiere introducir una corticoterapia. La búsqueda sistemática de anticuerpos antiplaquetas no se recomienda como primera elección para el diagnóstico de PTI [10] . Tanto en el niño como en el adulto, confirmar un diagnóstico de PTI requiere haber descartado una trombocitopenia constitucional. Una trombocitopenia constitucional debe considerarse ante toda trombocitopenia crónica, a fortiori en caso de trombocitopenia supuestamente autoinmunitaria pero resistente a las inmunoglobulinas intravenosas o a los corticoides. Una trombocitopenia constitucional poco sintomática puede evidenciarse de forma fortuita en la edad adulta. El diagnóstico positivo de una trombocitopenia constitucional se basa en una anamnesis y una exploración física orientadas hacia la búsqueda de signos extrahematológicos, así como en un análisis del frotis sanguíneo por un citólogo experimentado. La orientación etiológica de una trombocitopenia constitucional se basa en el carácter sindrómico o aislado de la trombocitopenia y el tamaño de las plaquetas en el frotis. La búsqueda de una trombocitopatía asociada se realiza sistemáticamente por medio del estudio de las funciones plaquetarias por agregometría (bajo reserva de un recuento plaquetario compatible con la realización de la prueba) y de la expresión de las glucoproteínas membranarias plaquetarias por citometría de flujo (CMF). Los demás exámenes varían según el contexto y la etiología sospechada: estudio de la ultraestructura plaquetaria por microscopia electrónica de transmisión, determinación de la TPO sérica, determinación del FVW incluyendo una determinación de la agregación de un plasma rico en plaquetas a diferentes concentraciones de ristocetina (RIPA: agregación plaquetaria inducida por ristocetina), mielograma, estudio radiológico óseo, análisis citogenéticos (cariotipo constitucional o técnica de fluorescencia in situ por hibridación [FISH] en busca de deleciones que afecten a las regiones 11q23-24 y 22q11-2 responsables respectivamente del síndrome de Jacobsen y del síndrome de DiGeorge), ecografías cardíaca y abdominal, determinación ponderal de las inmunoglobulinas, secuenciación de un gen candidato, etc. EMC - Tratado de medicina Hemostasia primaria E – 1-1272 Trombocitopenia (confirmada en citrato) Exploración física, hemograma, biología Argumentos a favor de un origen central Sí Mielograma No Trombocitopenia central Hemopatías malignas (leucemia, mielodisplasia) Trombocitopenia periférica Trombocitopenia por secuestro Trombocitopenia por consumo Trombocitopenia por hiperdestrucción Aplasia medular Transfusión masiva Coagulación intravascular diseminada2 Púrpura trombocitopénica inmunológica 4 Invasión metastásica Hiperesplenismo1 Microangiopatía trombótica3 Enfermedades autoinmunitarias 5 Carencia de vitaminas B9, B12 Megaloblastosis no carencial (alcohol, medicamentosa) Disglobulinemias 6 Infecciosas 7 Medicamentosas Aloinmunitarias 8 Figura 3. Árbol de decisiones. Diagnóstico de una trombocitopenia. 1. Signos de hipertensión portal, esplenomegalia, anomalías de las pruebas hepáticas; 2. tiempo parcial de tromboplastina activada y tiempo de protrombina anormales, ↓ fibrinógeno, ↑ dímeros D, ↑ monómeros de fibrina; 3. insuficiencia renal, hemólisis eritrocítica, presencia de esquistocitos en el frotis; 4. diagnóstico de eliminación que hay que considerar en caso de trombocitopenia aislada; 5. signos a favor de un lupus, síndrome de los antifosfolípidos, etc.; 6. búsqueda de gammapatía monoclonal por electroforesis de las proteínas séricas; 7. serologías de virus de las hepatitis B, C, virus de la inmunodeficiencia humana; 8. trombocitopenia neonatal por aloinmunización maternofetal, púrpura postransfusional. Exploración de una enfermedad de Von Willebrand La detección de una enfermedad de Von Willebrand se basa en la determinación de tres parámetros: el antígeno del FVW (FVW:Ag), la actividad de cofactor de la ristocetina (FVW:RCo) u otra prueba funcional similar que evalúe la unión FVW-GPIb y la actividad coagulante del FVIII (FVIII:C). El cálculo de las relaciones FVIII:C/FVW:Ag y FVW:RCo/FVW:Ag forma parte de la detección. La existencia de una trombocitopenia macrocítica y fluctuante en el hemograma orienta hacia una enfermedad de Von Willebrand de tipo 2B. Un TOP normal tiene un excelente valor predictivo negativo para el diagnóstico de enfermedad de Von Willebrand, excepto de la variante 2N, que se manifiesta por un déficit aislado de FVIII secundario a un defecto de la unión del FVW al FVIII. Se recomienda una RIPA en las etapas de detección, si está disponible. Este estudio de primera línea debe efectuarse a distancia de un síndrome inflamatorio o de un embarazo, que se asocian a un aumento fisiológico de la concentración de FVW (Fig. 4). La interpretación debe tener también en cuenta el grupo eritrocítico ABO, pues los individuos del grupo O tienen fisiológicamente concentraciones de FVW más bajas que los de los grupos no O. Una disminución proporcional de la concentración de FVW:Ag y FVW:RCo orienta hacia una enfermedad de Von Willebrand EMC - Tratado de medicina cuantitativa de tipo 1. En el tipo 2, el FVW:RCo está proporcionalmente más disminuido que la concentración de FVW:Ag (relación FVW:RCo/FVW:Ag < 0,7), lo cual indica una anomalía cualitativa. En el tipo 3, la concentración de FVW:Ag y FVW:RCo es indetectable y el FVIII es muy bajo. Un déficit aislado de FVIII:C puede deberse a una enfermedad de Von Willebrand de tipo 2N o a una forma atenuada de hemofilia A. El diagnóstico diferencial se basa entonces en el estudio de la unión del FVW al FVIII. Una agregación paradójica a bajas concentraciones de ristocetina en RIPA sugiere, o bien una enfermedad de Von Willebrand 2B (tratada con concentrados de Von Willebrand), o bien una seudoenfermedad de Von Willebrand plaquetaria (tratada con transfusiones de plaquetas), cuyo diagnóstico diferencial se basa en el estudio de los genes VWF y GP1BA. La caracterización fenotípica de una enfermedad de Von Willebrand, en particular de las variantes cualitativas (2A, 2B, 2M), requiere otros exámenes especializados (perfil multimérico, estudio de la unión del FVW al colágeno, determinación del propéptido) que no se detallan aquí [7] (Fig. 4). Exploración de una trombocitopatía Exploración de primera línea Como complemento del hemograma y del estudio del frotis sanguíneo, la International Society on Thrombosis and Hemostasis 5 E – 1-1272 Hemostasia primaria Sospecha de enfermedad de Von Willebrand 1 FVW:AG, FVW:RCO, FVIII: C ± TOP y RIPA 2 Anormal FVIII:C/FVW: Ag < 0,5–0,6 RIPA positivo 3 FVW:AG y FVW:RCO < 30% o FVW:RCO/FVW:Ag < 0,7 Enfermedad de Von Willebrand de tipo 2N o hemofilia A Enfermedad de Von Willebrand de tipo 2B o seudoenfermedad de Von Willebrand Enfermedad de Von Willebrand de tipo 1, 2 o 3 Figura 4. Árbol de decisiones. Diagnóstico de una enfermedad de Von Willebrand. FVW: factor de Von Willebrand; FVW:Ag: antígeno del factor de Von Willebrand; FVW:RCo: actividad de cofactor de la ristocetina; FVW:CB: estudio de la unión del factor de Von Willebrand al colágeno; FVIII:C: factor VIII coagulante; TOP: tiempo de obturación plaquetaria; RIPA (agregación plaquetaria inducida por ristocetina): medición de la agregación de un plasma rico en plaquetas a diferentes concentraciones de ristocetina. 1. En un individuo mayor de 50 años o en presencia de una neoplasia, una enfermedad autoinmunitaria o una gammapatía monoclonal: sospechar un síndrome de Von Willebrand adquirido; 2. un TOP-difosfato de adenosina normal tiene un buen valor predictivo negativo para la enfermedad de Von Willebrand (excepto la variante 2N); 3. presencia de una aglutinación paradójica a concentraciones bajas de ristocetina (< 0,8 mg/ml) en RIPA. Diagnóstico diferencial y tipificación Test unión FVW-FVIII Secuenciación FVW, GP1BA Perfil multimérico, propéptido FVW:CB, genotipificación, etc. (ISTH) recomienda la realización sistemática de las siguientes pruebas: un estudio de las funciones plaquetarias por agregometría óptica, un estudio de un marcador de secreción plaquetaria y un análisis de la expresión de las principales glucoproteínas membranarias plaquetarias por CMF (Fig. 5) [9] . La agregometría óptica es la prueba de referencia para la exploración de las funciones plaquetarias. Evalúa in vitro la agregación plaquetaria en presencia de inductores específicos. Este examen se realiza en un plasma rico en plaquetas (PRP) obtenido después de centrifugación de sangre total citratada a 250 g durante 10 minutos a temperatura ambiente. No se recomienda ajustar el recuento plaquetario en PRP excepto en caso de trombocitosis superior a 600 g/l [11] . Se utiliza un grupo limitado de agonistas como primera elección (adrenalina, ADP, colágeno, ácido araquidónico y ristocetina). La agregometría puede orientar de entrada el diagnóstico en caso de perfil característico (Cuadro 1) [8] . La presencia de un defecto de la agregación plaquetaria a todos los agonistas excepto a la ristocetina que contrasta con una expresión conservada de GPIIbIIIa (␣IIb3) en CMF orienta hacia un trastorno de la activación de la integrina ␣IIb3 secundario a una variante cualitativa de Glanzmann, un déficit de factor intercambiador de nucleótidos de guanina-1 regulado por diacilglicerol y calcio (CalDAG-GEF1) o un déficit de kindlina-3. Un perfil de agregación inespecífico induce a buscar una anomalía compleja de la señalización plaquetaria o una anomalía granular. Un panel más extenso es útil en segunda línea para ayudar a caracterizar la naturaleza del déficit [8] . A causa de la falta de sensibilidad de la agregometría óptica en las trombocitopatías de secreción, ahora se recomienda efectuar el estudio de un marcador de los gránulos densos (ATP, serotonina) y de un marcador de secreción de los gránulos ␣ (P-selectina) [8, 9] . La CMF permite una cuantificación de las principales glucoproteínas membranarias plaquetarias en reposo o después de activación. Es útil en el niño para el diagnóstico del SBS y la TG, porque sólo requiere una pequeña cantidad de sangre. También permite una exploración indirecta de los gránulos densos y ␣. Exámenes especializados A veces, es necesaria una exploración compleja, en laboratorios especializados, para el diagnóstico de una trombocitopatía. Ante el coste y la disponibilidad restringida de estos exámenes, el empleo sistemático de un cuestionario estandarizado que permita el cálculo de un índice hemorrágico dotado de un buen 6 valor predictivo negativo podría permitir dirigir mejor estas investigaciones [12] . La medición de la serotonina y la determinación de los nucleótidos intraplaquetarios por cromatografía líquida de alto rendimiento (HPLC) son pruebas sensibles a las anomalías de los gránulos densos, pero de disponibilidad muy restringida comparada con la medición de la secreción de ATP por lumiagregometría [13] . La microscopia electrónica, que permite un estudio de la ultraestructura plaquetaria, es el examen de referencia para confirmar un déficit cuantitativo de gránulos ␣ o (Fig. 5). En caso de fenotipo sugestivo, se realiza la secuenciación de uno o varios genes candidatos. Si no se encuentra una mutación candidata, puede proponerse un enfoque de tipo «secuenciación del exoma completo» por secuenciación de alto flujo, bajo reserva de una familia suficientemente informativa para establecer la distinción entre las variantes encontradas entre mutaciones candidatas y simples polimorfismos [11] . Etiologías de los trastornos de la hemostasia primaria Trombocitopenia Trombocitopenia adquirida La trombocitopenia suele ser de origen adquirido. Después de descartar una seudotrombocitopenia, se distinguen dos grandes grupos etiológicos: las trombocitopenias centrales secundarias a un trastorno de producción medular (en un contexto de hemopatía maligna o no) y las trombocitopenias periféricas por destrucción, consumo o secuestro de las plaquetas circulantes. Las trombocitopenias por secuestro se observan en las enfermedades asociadas a una esplenomegalia (hepatopatías con hipertensión portal, hemopatías, enfermedad de sobrecarga). Las trombocitopenias por consumo reúnen las microangiopatías trombóticas y la coagulación intravascular diseminada (CIVD). Entre las etiologías de trombocitopenia periférica asociadas a una hiperdestrucción plaquetaria, la PTI representa la etiología más frecuente. El diagnóstico de PTI sólo puede sospecharse en caso de trombocitopenia aislada después de descartar otras causas de trombocitopenia. Pueden observarse causas específicas de trombocitopenia en un contexto neonatal (trombocitopenia por aloinmunización maternofetal), obstétrica (trombocitopenia gestacional, preeclampsia, EMC - Tratado de medicina Hemostasia primaria E – 1-1272 “ Punto importante “ Punto importante Argumentos diagnósticos a favor de una trombocitopenia constitucional • Anamnesis: – antecedentes familiares hemorrágicos o de trombocitopenia; – antecedentes familiares de hemopatía mieloide o de mielodisplasia; – inicio precoz de las manifestaciones hemorrágicas (período neonatal). • Exploración física: trombocitopenia asociada a signos extrahematológicos: – infecciones de repetición, eccema; – síndrome malformativo (aplasia radial, retraso mental, etc.); – sordera, catarata o afectación renal; – xantomas tendinosos; – albinismo. • Hemograma y frotis sanguíneo: – persistencia de una trombocitopenia estable desde hace años; – tamaño o morfología anormales de las plaquetas en el frotis sanguíneo (microplaquetas, macroplaquetas, plaquetas grises); – morfología leucocítica anormal (inclusiones leucocíticas: cuerpos de Döhle); – morfología eritrocítica anormal (microcitosis, macrocitosis, estomatocitosis, poiquilocitosis, dacriocitos). • Respuesta a los tratamientos inmunomoduladores y transfusionales: – ausencia de respuesta a las inmunoglobulinas polivalentes por vía intravenosa (IgIV) o a los corticoides; – respuesta a las transfusiones plaquetarias. Detección de una enfermedad de Von Willebrand • Se basa en la determinación de tres parámetros (FVW:Ag, FVW:RCo y FVIII:C) y el cálculo de las relaciones FVIII:C/FVW:Ag y FVW:RCo/FVW:Ag. • Un TOP normal tiene un buen valor predictivo negativo para el diagnóstico de enfermedad de Von Willebrand constitucional (excepto la variante 2N) o adquirida. renciación megacariocítica, la maduración megacariocítica o la trombopoyesis [14] . Actualmente, sólo se identifica una anomalía molecular en el 50% de los casos de trombocitopenia constitucional (Cuadro 2). Trombocitopenia constitucional sindrómica Microcítica. El síndrome de Wiskott-Aldrich (SWA) se debe a una anomalía de la proteína SWA, que regula la polimerización de la actina en las células hematopoyéticas. Se caracteriza Sospecha de trombocitopatía Exploración de 1.ª línea Exploración especializada Prueba de la mepacrina (citometría de flujo) Microscopia electrónica Microscopia confocal Determinación de TPO sérica Western blot en lisado plaquetario, secuenciación, gen candidato Secuenciación del exoma/ genoma completo Hemograma + frotis sanguíneo Agregometría plaquetaria con panel ampliado Estudio de un marcador de los gránulos densos 1 Citometría de flujo (expresión GPIIbIIIa, GPIb/IX/V, GPVI, P-selectina) síndrome de hemólisis, elevación de las enzimas hepáticas y recuento de plaquetas bajo [HELLP]) o transfusional (púrpura postransfusional). Figura 5. Árbol de decisiones. Jerarquización de los exámenes complementarios útiles para el diagnóstico de trombocitopatía. Diagnóstico de un trastorno de la hemostasia primaria. TPO: trombopoyetina; GP: glucoproteína. 1. Ejemplo: medición de la secreción de trifosfato de adenosina. Trombocitopenia constitucional Las trombocitopenias constitucionales reúnen un conjunto heterogéneo de anomalías genéticas que interfieren con la dife- Cuadro 1. Perfiles característicos de trombocitopatías en agregometría óptica con el panel básico de agonistas plaquetarios. ADP (M) 10 5 2,5 Adrenalina (M) Colágeno (g/ml) AA (mM) TRAP (M) Ristocetina (mg/ml) 5 5 1 50 1,5 1 SBS N N N N N/I A Seudoenfermedad de Von Willebrand y enfermedad de Von Willebrand 2B N N N N N AoI TG de tipo 1 (completa) a A A A A A NoI Déficit de CalDAG-GEFI N/I I I N/I N/I N Déficit de LAD-III A A A A I N Déficit de gránulos ␣ b N N N/I N N/I N Alteración de la secreción de los gránulos densos b N N/I N/I I/A I N N Anomalía del receptor GPVI N N A A N N N Déficit «de tipo aspirina» I I/A N/I I/A I/A N N Anomalía del receptor P2Y12 A/I N/I N/I I/A N/I N N I N/I A/I A I A/I I 0,75 ↑ ADP: difosfato de adenosina; AA: ácido araquidónico; TRAP: péptido activador del receptor de trombina; SBS: síndrome de Bernard-Soulier; TG: trombastenia de Glanzmann; LAD-III: alteración de la adhesión de los leucocitos de tipo III; A: ausencia de agregación; I: agregación intermedia o reversible; N: agregación normal; CalDAG-GEF1: factor intercambiador de nucleótidos de guanina-1 regulado por diacilglicerol y calcio; GP: glucoproteína. a Anomalías menos graves en caso de variante TG. b Anomalías inconstantes. EMC - Tratado de medicina 7 E – 1-1272 Hemostasia primaria Cuadro 2. Trombocitopenias constitucionales: anomalía genética causal y modo de transmisión. Trombocitopenias microcíticas Trombocitopenias normocíticas Trombocitopenias macrocíticas Trombocitopenia constitucional Transmisión Gen anormal (localización cromosómica) SWA XLT Ligada a X SWA (Xp11) Trombocitopenias con aplasia radial AR RBM8A (1q21.1) Síndrome IVIC AD SALL4 (20q13.2) Amegacariocitosis con sinostosis radiocubital AD HOXA11 (7p15.2) Amegacariocitosis congénita (CAMT) AR c-Mpl (1p34) Trombocitopenia FPD/AML AD RUNX-1 (21q22) Trombocitopenia ANKRD26 AD ANKRD26 (10p2) Trombocitopenia ETV6 AD ETV6 (12p13) Trombocitopenia con mutación del citocromo C AD CYCS (7p15.3) Trombocitopenia de Québec AD PLAU (10q22.2) Síndromes MYH9 AD MYH9 (22q12.13) Trombocitopenia de Paris-Trousseau/Jacobsen AD FLI-1 (deleción 11q23-24) Síndrome de DiGeorge AD GP1BB (deleción 22q11.2) Síndrome de Bernard-Soulier AR GP1BA (17p13.2), GP1BB (22q11), GP9 (3q21) Síndrome de las plaquetas grises AR NBEAL2 (3p21.1) Trombocitopenia con mutación de GFl1B AD GFl1B (9q34.13) Trombocitopenia mediterránea AD GP1BA (17p13) Seudoenfermedad de Von Willebrand plaquetaria AD GP1BA (17p13) Trombocitopenia ligada a X con diseritropoyesis (XLT) o síndrome talasémico (XLTT) Ligada a X GATA-1 (Xp11) Trombocitopenia TUBB1 AD TUBB1 (6p21.3) Trombocitopenia ACTN1 AD ACTN1 (14q24.1) Filaminopatías Ligada a X FLNA (Xq28) Trombocitopenia PRKACG AR PRKACG (9q21.11) Sitosterolemia hereditaria AR ABCG5, ABCG8 (2p21) AR: autosómica recesiva; AD: autosómica dominante; SWA: síndrome de Wiskott-Aldrich; FPD-AML: trombocitopenia familiar con predisposición a las leucemias. “ Punto importante Detección de una trombocitopatía • Un TOP normal no permite descartar una trombocitopatía. • La agregometría óptica constituye el examen de referencia para el diagnóstico de una trombocitopatía. • Se recomienda el estudio de un marcador de los gránulos densos (ATP, serotonina) y de un marcador de secreción de los gránulos ␣ (P-selectina) en caso de sospecha de trombocitopatía moderada, incluso cuando la agregometría óptica es normal. por un síndrome hemorrágico precoz y grave, una microtrombocitopenia casi patognomónica y una trombocitopatía del tipo de la enfermedad de los reservorios delta (dSPD). El cuadro completo comporta un eccema y un déficit inmunitario grave [15] . La terapia génica podría transformar el pronóstico del SWA, con riesgo de complicaciones autoinmunitarias y de hemopatías malignas. Normocítica. La trombocitopenia con aplasia radial (TAR) es una trombocitopenia grave (20-30 g/l), de aparición neonatal, de origen central, asociada a una aplasia radial bilateral que no afecta a los pulgares. La trombocitopenia se atenúa con la edad. A veces, existen otras alteraciones óseas o cardíacas. La anemia de Fanconi, que asocia una pancitopenia que empeora progresivamente y una aplasia de los pulgares, constituye un diagnóstico diferencial. El modo de transmisión genética de la TAR es original, porque resulta de la pérdida de un alelo del gen RBM8A por deleción (1q21.1) y de la presencia de un polimorfismo raro en el otro alelo [16] . 8 La amegacariocitosis con sinostosis radiocubital se debe a mutaciones del gen HOXA11, que codifica un factor de transcripción implicado en la morfogénesis del antebrazo y la megacariopoyesis. La sinostosis radiocubital responsable de una limitación de la pronosupinación es patognomónica. Puede evolucionar hacia la aplasia medular. El síndrome oculootorradial asocia una afectación de los músculos oculomotores, una sordera mixta, anomalías óseas y una trombocitopenia grave. Macrocítica. El síndrome de Jacobsen y su variante, la trombocitopenia de Paris-Trousseau, se deben a una deleción del brazo largo del cromosoma 11 (11q23.3-24.2), de tamaño variable, que implica una copia del gen friend leukemia integration 1 (FLI-1) esencial para la diferenciación megacariocítica. El síndrome de Jacobsen tiene una prevalencia estimada en un uno por 100.000. Suele aparecer de novo y se transmite de forma autosómica dominante. El complejo FLI-1/RUNX-1 permite al megacariocito iniciar su fase de poliploidización, reprimiendo la expresión de una proteína del citoesqueleto: la cadena pesada de la miosina IIB (MYH10). El síndrome de Jacobsen y la trombocitopenia de Paris-Trousseau se caracterizan por una macrotrombocitopenia moderada (30-80 g/l), a menudo asociada a una trombocitopatía relacionada con una anomalía de los gránulos ␣. A veces, se observa un gránulo ␣ gigante patognomónico al microscopio. En el caso del síndrome de Jacobsen, también existe un retraso de crecimiento ponderoestatural, un déficit intelectual moderado, un síndrome dismórfico que afecta a la cara y las extremidades y malformaciones viscerales, en particular cardíacas (comunicación interauricular o interventricular, coartación de aorta), genitourinarias (hipospadias, duplicación ureteral) o cerebrales [17] . La persistencia de la expresión intraplaquetaria de la proteína MYH10 constituye un argumento diagnóstico inespecífico. El cariotipo constitucional puede ser poco sensible para las deleciones de pequeño tamaño, cuya detección puede requerir el empleo de una técnica de fluorescencia in situ por hibridación. EMC - Tratado de medicina Hemostasia primaria E – 1-1272 El síndrome de DiGeorge es frecuente (uno de cada 4.000 nacimientos) y se manifiesta por un síndrome polimalformativo velocardiofacial de penetrancia variable, incluso dentro de una misma familia. La forma clásica asocia dismorfias faciales, malformaciones cardíacas, hipoplasia del timo y de las glándulas paratiroideas, que producen, respectivamente, un déficit inmunitario y una hipocalcemia. Se acompaña de una macrotrombocitopenia moderada (> 100 g/l) y asintomática. Se parece a una forma heterocigótica del SBS, porque está causada por una deleción del brazo largo del cromosoma 22 (22q11.2) que implica al locus que codifica la GPIb, elemento constitutivo del complejo GPIb-IX-V [18] . Las filaminopatías y la sitosterolemia hereditaria constituyen otras dos causas raras de trombocitopenia macrocítica sindrómica. Cuadro 3. Clasificación de la enfermedad de Von Willebrand constitucional (según Sadler et al [25] ). Tipo y subtipos Descripción Tipo 1 Déficit cuantitativo parcial de FVW Tipo 2 Déficit cualitativo de FVW 2A Alteración de la interacción FVW/plaquetas secundaria a la ausencia de MHPM 2B Aumento de la interacción FVW/plaquetas 2M Alteración de la interacción del FVW con plaquetas o colágeno no ligada a una anomalía de los MHPM 2N Alteración de la interacción FVW/FVIII Tipo 3 Trombocitopenia constitucional no sindrómica Microcítica. La trombocitopenia ligada a X es una forma atenuada del SWA sin eccema ni déficit inmunitario. Normocítica. La amegacariocitosis congénita se debe a mutaciones que conducen a una ausencia de expresión del receptor Mpl o a un receptor todavía expresado pero hipofuncional. Se manifiesta por hemorragias precoces, a veces graves (hemorragia intracerebral, digestiva), correlacionadas con la intensidad de la trombocitopenia (< 30 g/l) presente desde el nacimiento. Evoluciona espontáneamente hacia la aplasia medular. La concentración de TPO sérica es muy elevada, lo cual traduce la disminución importante de la masa megacariocítica. El único tratamiento curativo es el trasplante de médula [19] . Una trombocitopenia normocítica, no sindrómica y de transmisión autosómica dominante induce a buscar tres etiologías de trombocitopenia constitucional con riesgo de hemopatías malignas. La trombocitopenia por trastorno plaquetario familiar con predisposición a la leucemia mieloide aguda (FPD/AML) se debe a una mutación del factor de transcripción RUNX-1, que, en el complejo del factor de unión nuclear (CBF), regula la diferenciación y la proliferación de las células madre hematopoyéticas. El recuento plaquetario suele estar poco disminuido e incluso puede ser normal. A veces, se asocia a una trombocitopatía de tipo ␦-SPD. Existe una susceptibilidad aumentada a las hemopatías mieloides, por las mutaciones con efecto dominante negativo [20] . La trombocitopenia familiar autosómica dominante ligada al cromosoma 10 (thrombocytopenia-2 [THC2]) se debe a una mutación del promotor del gen ANKRD26, causante de un fallo en la unión de los factores de transcripción FLI-1 y RUNX-1, que reprimen la expresión del ANKRD26 durante la fase tardía de la megacariopoyesis. La trombocitopenia es moderada, pero algunas mutaciones se asocian a una trombocitopenia por disminución del contenido en gránulos ␣. La THC2 predispone al desarrollo de una leucemia mieloide aguda [21] . La afectación del gen ETV6 (ETS variante 6) da lugar a una trombocitopenia con macrocitosis eritrocítica y predispone al desarrollo de una leucemia linfoide aguda [22] . Macrocítica. La mayoría de las macrotrombocitopenias hereditarias se deben a una anomalía de la maduración megacariocítica o de la formación de las proplaquetas. El síndrome de la cadena pesada de la miosina-9 (MYH9) es una de las causas más frecuentes de trombocitopenia constitucional [23] . Está constituido por varias entidades clínico-biológicas relacionadas con mutaciones del gen MYH9, que codifica la cadena pesada de la miosina no muscular de tipo IIA, esencial para las funciones contráctiles y secretoras de la plaqueta. El síndrome hemorrágico es moderado, incluso está ausente. La macrotrombocitopenia puede ser aislada o asociarse a una nefropatía, una sordera de percepción o una catarata, en función de la localización de la mutación causal. Estas manifestaciones extrahematológicas pueden aparecer secundariamente a la trombocitopenia. La visualización de macroplaquetas y de inclusiones leucocíticas basófilas (seudocuerpos de Döhle) en el frotis sanguíneo es sugestiva. La genotipificación confirma el diagnóstico. Una trombocitopenia macrocítica también puede indicar un trastorno de la trombopoyesis secundario a una anomalía de una proteína del citoesqueleto (β1-tubulina, α-actinina o diaphanous-related formin 1 [DIAHP1]) o implicada en una vía de señalización [24] . EMC - Tratado de medicina Déficit cuantitativo total de FVW FVW: factor de Von Willebrand; MHPM: multímeros de alto peso molecular del FVW; FVIII: factor VIII. La trombocitopenia macrocítica ligada a X con diseritropoyesis (XLT) o con síndrome talasémico (XLTT) son dos entidades de trombocitopenia constitucional macrocítica secundarias a mutaciones del factor de transcripción GATA-1. Puede observarse una disminución de la agregación al colágeno en la agregometría [16] . La variante 2B de enfermedad de Von Willebrand y ciertas etiologías de trombocitopatías constitucionales (SBS, seudoenfermedad de Von Willebrand plaquetaria, síndrome de las plaquetas grises) también se caracterizan por una trombocitopenia macrocítica. Enfermedad de Von Willebrand La enfermedad de Von Willebrand es una enfermedad hemorrágica que se debe a un déficit cuantitativo o cualitativo de FVW. Suele ser de origen constitucional, pero también existen raras formas adquiridas. Enfermedad de Von Willebrand constitucional La prevalencia de las formas sintomáticas de enfermedad de Von Willebrand se estima en un 0,01% de la población. Se distinguen tres tipos de enfermedad de Von Willebrand según la naturaleza del déficit: cuantitativo parcial (tipo 1), cualitativo (tipo 2) o cuantitativo completo (tipo 3). El tipo 2 reúne cuatro subtipos: 2A, 2B, 2M y 2N. La transmisión del déficit suele tener lugar de modo autosómico dominante, excepto en los tipos 3, 2N y excepcionales variantes 2A de transmisión recesiva (Cuadro 3). La enfermedad de Von Willebrand se manifiesta por hemorragias cutaneomucosas espontáneas o posoperatorias, según la intensidad del déficit. Las formas más graves asociadas a un déficit grave de FVIII se complican también con hematomas y hemartrosis. El tratamiento de la enfermedad de Von Willebrand se basa en la administración por vía intravenosa o intranasal de desmopresina en los individuos buenos respondedores comprobados, lo cual ocurre en la mayoría de los tipos 1. En caso de contraindicación (variante 2B y tipo 3) o de respuesta insuficiente a la desmopresina (la mayoría de los tipos 2), el tratamiento se basa en la administración intravenosa de concentrados de factor de Von Willebrand purificado [7] . Síndrome de Von Willebrand adquirido El síndrome de Von Willebrand adquirido tiene la misma expresión clínico-biológica que la enfermedad de Von Willebrand constitucional. Se sospecha ante la coexistencia de un déficit de FVW y hemorragias cutaneomucosas de aparición reciente en un individuo a menudo anciano, sin antecedentes hemorrágicos personales o familiares. El estudio etiológico busca una enfermedad subyacente susceptible de interferir con la síntesis, la proteólisis, el aclaramiento o la función del FVW (disglobulinemias monoclonales, valvulopatías cardíacas, neoplasias, hemopatías). El tratamiento de la enfermedad subyacente (inmunosupresores, quimioterapia, recambio valvular, etc.), cuando es posible, permite obtener la remisión del síndrome de Von Willebrand adquirido. En cuanto al tratamiento hemostático, 9 E – 1-1272 Hemostasia primaria las opciones terapéuticas (desmopresina, concentrados de FVW o de FVIII, inmunoglobulinas polivalentes) difieren según la etiología [7] . Trombocitopatías Trombocitopatías adquiridas Las trombocitopatías medicamentosas constituyen la primera causa de trombocitopatía. El ácido acetilsalicílico y los antiinflamatorios no esteroideos (AINE), inhibidores de la ciclooxigenasa de tipo 1 (COX-1), y los inhibidores de P2Y12 (clopidogrel, ticagrelor, prasugrel) son los causantes más frecuentes. Algunos inhibidores de la tirosina-cinasa utilizados como antitumorales aumentan el riesgo hemorrágico al interferir con ciertas vías de señalización intraplaquetaria [26] . Una trombocitopatía adquirida puede complicar la evolución de una hemopatía según diferentes mecanismos: autoanticuerpos anti-GPIb o anti-GPIIbIIIa, trombocitopatía ␦-SPD secundaria a una dismegacariopoyesis. Puede observarse una separación del ectodominio de GPVI en presencia de autoanticuerpos o por la acción de metaloproteasas plaquetarias. La trombocitopatía asociada a la insuficiencia renal parece deberse principalmente a la disminución del hematocrito por disminución de la síntesis renal de eritropoyetina (EPO). Los glóbulos rojos son una fuente de ADP, un potente inductor de la agregación plaquetaria, y contribuyen en el aspecto reológico a la adhesión plaquetaria, al mantener las plaquetas en la periferia del vaso, favoreciendo así su interacción con la pared vascular cuando está lesionada. Trombocitopatías constitucionales Las trombocitopatías constitucionales reúnen un conjunto heterogéneo de enfermedades hemorrágicas raras que interfieren con una o varias funciones plaquetarias, según la localización de la anomalía [27] . Anomalías de los receptores membranarios de adhesión Complejo GPIb-IX-V. El SBS, con una prevalencia estimada de 1 por 106 nacimientos, depende de un déficit cuantitativo o más raramente cualitativo del complejo GPIb-IX-V de transmisión autosómica recesiva. Asocia una trombocitopenia macrocítica y una trombocitopatía por alteración de la adhesión plaquetaria al FVW relacionada con el déficit de GPIb␣. Se manifiesta en la infancia por una púrpura cutaneomucosa que suele ser grave. El diagnóstico se basa en la existencia de una trombocitopenia con plaquetas gigantes (de tamaño superior a un hematíe) asociada a una disminución aislada de la aglutinación de las plaquetas con ristocetina (Cuadro 1). El diagnóstico se confirma mediante el estudio cuantitativo de GPIbα en CMF y la biología molecular. Algunas formas monoalélicas de SBS secundarias a mutaciones de GPIbα se manifiestan por una macrotrombocitopenia benigna, aislada, transmitida de modo autosómico dominante (macrotrombocitopenia mediterránea) o recesiva (variante Bolzano). También se describen formas homocigóticas excepcionales de SBS en pacientes portadores a la vez de una microdeleción 22q11.2 responsable de un síndrome de DiGeorge (deleción que incluye un alelo de GPIbβ) y una mutación de GPIbβ en el alelo contralateral sin deleción. La seudoenfermedad de Von Willebrand plaquetaria se debe a una mutación del gen GPIBA, que aumenta la afinidad de la GPIbα por el FVW. Esta trombocitopatía constituye la variante de la enfermedad de Von Willebrand de tipo 2B, también caracterizada por una mayor interacción FVW-GPIbα relacionada con una mutación en el exón 28 del gen FVW. La expresión clínica de estas dos enfermedades es idéntica, asocian una macrotrombocitopenia fluctuante y una agregación plaquetaria paradójica a bajas concentraciones de ristocetina (≤ 0,8 mg/ml). El diagnóstico diferencial se basa ahora en la secuenciación de los genes GPIBA y FVW. Receptor GPVI. Una disminución aislada de la agregación plaquetaria al colágeno o a sus análogos orienta hacia un déficit constitucional o adquirido de GPVI o de la vía de señalización posterior a este receptor (Cuadro 1). Sólo se han descrito algunos casos de déficit constitucional, con un síndrome hemorrágico moderado. 10 Anomalías de los receptores de los agonistas solubles Receptores purinérgicos. Las plaquetas expresan en su membrana tres receptores purinérgicos. El P2Y1 y el P2Y12 son receptores acoplados a las proteínas G (RCPG) que se unen al ADP, mientras que el receptor P2X1 se une al ATP y tiene una función de canal cálcico. La afectación del receptor P2Y12 se acompaña de una alteración de la agregación al ADP in vitro, sea cual sea la dosis empleada, y de una respuesta disminuida de las plaquetas a dosis bajas de otros agonistas. En la gran mayoría de los casos, estas anomalías reflejan la toma de un antiagregante plaquetario dirigido al P2Y12 (clopidogrel, prasugrel, cangrelor, ticagrelor, etc.). Al contrario que el P2Y1, existen raros casos de anomalías constitucionales del receptor P2Y12 responsables de un síndrome hemorrágico moderado. Sólo se ha descrito un caso de déficit constitucional de P2X1, que se manifiesta por una anomalía aislada de la agregación plaquetaria al ADP. Receptor del tromboxano A2. Una anomalía de la vía del TXA2 se manifiesta por una agregación ausente a dosis elevadas de ácido araquidónico y disminuida a bajas concentraciones de numerosos agonistas, como el ADP y la adrenalina (Cuadro 1). Indica excepcionalmente una trombocitopatía constitucional por alteración de la síntesis del TXA2 o por alteración de la respuesta al TXA2 a causa de una anomalía del receptor TPα o de la vía de señalización posterior. Este perfil de agregación refleja casi siempre una alteración de la conversión del ácido araquidónico en TXA2 relacionada con la toma de ácido acetilsalicílico o de un AINE inhibidor de la COX-1. Anomalías de las vías se señalización La identificación de las trombocitopatías ligadas a una alteración de la señalización plaquetaria es compleja, a juzgar por los raros casos descritos [11, 24] , pero este campo progresará gracias al impulso de las técnicas de secuenciación de alto flujo, como ilustra el descubrimiento reciente de una mutación con aumento de función de Src, causante de un síndrome de transmisión autosómica dominante que asocia afectación plaquetaria, mielofibrosis y anomalías óseas [28] . Anomalías de los gránulos plaquetarios Estas trombocitopatías se deben a una alteración de síntesis, almacenaje o secreción de los gránulos intraplaquetarios (gránulos ␣ y gránulos densos). Déficit de gránulos ␣. El síndrome de las plaquetas grises (SPG) es una trombocitopatía rara de transmisión autosómica recesiva, debida a mutaciones del gen neurobeachin-like 2 (NBEAL2), caracterizada por una trombocitopenia macrocítica de intensidad variable con una morfología plaquetaria característica en el frotis [16] . A veces, se observa una esplenomegalia o una mielofibrosis. La concentración sérica de vitamina B12 está aumentada. El examen de las funciones plaquetarias muestra un perfil variable según los pacientes (disminución de la agregación a la trombina y al colágeno), pero que puede ser normal (Cuadro 1). La microscopia electrónica es el examen de referencia para objetivar el déficit de gránulos ␣. Un déficit de gránulos ␣ también está presente en enfermedades distintas del SPG: • el síndrome de artrogriposis-insuficiencia renal-colestasis (síndrome ARC), de transmisión autosómica recesiva; • la trombocitopenia recesiva ligara a X por mutación de GATA-1; • y una trombocitopatía de transmisión autosómica dominante, por mutación del gen que codifica el factor de transcripción factor de crecimiento independiente 1B (GFl1B) [16] . El síndrome de Québec es otro ejemplo de trombocitopatía por anomalía del contenido de los gránulos ␣. Se debe a la sobreexpresión en los gránulos ␣ del activador del plasminógeno de tipo urocinasa (u-PA) causada por una duplicación en tándem del gen PLAU. La trombocitopenia es moderada o está ausente. La expresión clínica de esta trombocitopatía es poco frecuente, con sangrados esencialmente de naturaleza provocada que aparecen de manera retardada después de un traumatismo o un acto quirúrgico. El tratamiento se basa en la administración de antifibrinolíticos [15] . EMC - Tratado de medicina Hemostasia primaria E – 1-1272 Déficit de gránulos densos. Se sospecha un déficit de gránulos densos () o una anomalía de secreción ante una disminución de la agregación plaquetaria con bajas concentraciones de ADP, colágeno y adrenalina (Cuadro 1). Este perfil indica la importancia del ADP en la amplificación y el mantenimiento de la activación plaquetaria. Sin embargo, la agregometría tradicional carece de sensibilidad, lo cual justifica, durante la exploración inicial de un individuo con una posible trombocitopatía de secreción, el estudio de un marcador de secreción de los gránulos densos como el ATP, cuya determinación puede efectuarse fácilmente por quimioluminiscencia [9] . En caso de anomalía, la microscopia electrónica es el examen de referencia para evidenciar un déficit cuantitativo total o parcial de gránulos densos. El síndrome hemorrágico suele ser moderado. La presencia de un albinismo hace sospechar dos formas raras sindrómicas de déficit de gránulos : los síndromes de HermanskyPudlak y Chediak-Higashi. Las formas no sindrómicas de déficit de gránulos , o bien por anomalía de almacenaje (enfermedad de los reservorios delta [dSPD]), o bien por anomalía de secreción (λ-secretion defect), son causas frecuentes de trombocitopatía constitucional, aunque la anomalía molecular subyacente sólo se conoce en casos muy raros [28] . Anomalías de la función procoagulante plaquetaria El síndrome de Scott, de transmisión autosómica recesiva, se debe a una alteración de la activación de la proteína transmembranaria escramblasa (TMEM), que permite la exposición de los fosfolípidos aniónicos plaquetarios necesarios para la captación de los factores de coagulación en la membrana de la plaqueta activada. Es responsable de hemorragias posoperatorias. El recuento, la morfología plaquetaria, el TOP y la agregación plaquetaria son normales. La CMF es sugestiva en caso de disminución de la expresión de la anexina V después de activación plaquetaria. El síndrome de Stormorken se caracteriza por un aumento de la externalización de la fosfatidilserina. Es de transmisión autosómica dominante y se manifiesta paradójicamente por un fenotipo hemorrágico moderado, asociado a una ictiosis cutánea, una asplenia y una afectación neurológica [16] . Anomalías de la agregación plaquetaria Trombastenia de Glanzmann y sus variantes. La TG, de transmisión autosómica recesiva, se debe a una disminución de la expresión del complejo ␣IIb3 en la superficie de las plaquetas, que da lugar a una alteración de la agregación a todos los agonistas, excepto a la ristocetina (Cuadro 1). El recuento y la morfología plaquetaria suelen ser normales, pero algunas formas se asocian a una ligera macrotrombocitopenia. El diagnóstico se confirma por una alteración de la expresión de GPIIbIIIa en CMF. El tratamiento se basa en la transfusión de concentrados de plaquetas, cuya eficacia puede verse comprometida en caso de aloinmunización antiantígeno leucocítico humano (anti-HLA) o de isoinmunización anti-GPIIbIIIa, en cuyo caso se requiere un tratamiento con factor VII activado recombinante. Las variantes cualitativas de TG se asocian a mutaciones que suelen afectar a ITGB3, causantes de una alteración de la activación de ␣IIb3, que impide su unión al fibrinógeno. Los pacientes con una variante de TG se caracterizan pues por una alteración de la agregación plaquetaria, lo cual los diferencia de los portadores sanos heterocigóticos de TG, cuyas plaquetas expresan un nivel intermedio de GPIIbIIIa sin anomalías asociadas de la función plaquetaria [16] . Déficit de CalDAG-GEFI. La CalDAG-GEFI es una proteína intraplaquetaria implicada en la vía de señalización dentro-fuera que permite la activación de la ␣IIb3. Las mutaciones del gen RASGRP2 asociadas a un déficit cuantitativo o cualitativo de CalDAG-GEFI se manifiestan por una trombocitopatía moderada a grave, de transmisión autosómica recesiva. El recuento y el volumen plaquetario son normales. El TOP está muy prolongado. Las anomalías presentes en agregometría se parecen a las alteraciones de la vía P2Y12/Gi a causa de la importancia del ADP en la activación de CalDAG-GEFI. En presencia de dosis bajas de ADP o de péptido activador del receptor de trombina (TRAP), la agregación plaquetaria está muy disminuida. Esta alteración de la agregación se atenúa cuando estos dos agonistas se utilizan a dosis elevadas EMC - Tratado de medicina (Cuadro 1). La CMF objetiva una alteración de la activación de la ␣IIb3 [11] . Déficit de kindlina-3. La alteración de la adhesión de los leucocitos de tipo III (LAD-III), de transmisión autosómica recesiva, asocia un síndrome hemorrágico grave y una inmunodepresión. Este fenotipo muestra la importancia funcional de la kindlina-3, proteína submembranaria esencial para la activación de la integrina ␣IIb3 plaquetaria y de las -integrinas leucocíticas [16] . Anomalías vasculares Anomalías hereditarias del tejido conjuntivo Las enfermedades hereditarias del tejido conjuntivo pueden manifestarse por un síndrome hemorrágico cutaneomucoso [29] . La exploración física busca signos de orientación: hiperlaxitud cutaneoligamentosa (enfermedad de Ehlers-Danlos), telangiectasias (enfermedad de Rendu-Osler) o lesiones cutáneas (seudoxantoma elástico). Enfermedades vasculares adquiridas Vasculitis infecciosas Una púrpura febril puede indicar una vasculitis infecciosa, con un cuadro de púrpura fulminante al extremo. Una púrpura febril también puede revelar una endocarditis de Osler y motivar la realización de hemocultivos y de una ecografía cardíaca. Vasculitis inmunoalérgicas o por depósitos de complejos inmunitarios Una púrpura vascular puede marcar la evolución de una gammapatía monoclonal con o sin amiloidosis o de una hipergammaglobulinemia policlonal plasmática. La topografía de la púrpura es sugestiva en caso de amiloidosis, con lesiones purpúricas localizadas en los párpados, el cuello y los pliegues. Una púrpura que evoluciona por accesos desencadenados por la exposición al frío en un contexto de neuropatía periférica o de glomerulopatía hace sospechar una púrpura crioglobulinémica, que puede complicar la evolución de una hepatitis C o de un síndrome linfoproliferativo. La tríada de púrpura vascular ortostática, artralgias y afectación renal (hematuria, insuficiencia renal) en un niño o un adulto joven orienta hacia una púrpura reumatoidea. También puede observarse una púrpura vascular en ciertas vasculitis sistémicas o conectivitis autoinmunitarias. Causa carencial: el escorbuto El escorbuto es una causa rara de púrpura (púrpura petequial perifolicular y equimótica, gingivorragias, descarnadura dental) identificada por una determinación de la vitamina C. Causas atróficas La púrpura atrófica es un diagnóstico de eliminación. Se sospecha en caso de púrpura localizada en la cara dorsal de las manos y los antebrazos. Se observa en el anciano o después de una corticoterapia prolongada. Conclusión El diagnóstico de los trastornos de la hemostasia primaria, en particular de las enfermedades plaquetarias hereditarias, está a punto de sufrir una revolución debido a la llegada de las nuevas técnicas de secuenciación de alto flujo. Estas técnicas deberían permitir próximamente la secuenciación simultánea de un amplio panel de genes candidatos [30] e identificar los polimorfismos genéticos que modulan el fenotipo hemorrágico. Bibliografía [1] [2] [3] Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost 2016;14:918–30. Springer TA. Von Willebrand factor, Jedi knight of the blood stream. Blood 2014;124:1412–25. Deutsch VR, Tomaer A. Advances in megacaryocytopoiesis and thrombopoiesis: from bench to bedside. Br J Haematol 2013;161:778–93. 11 E – 1-1272 Hemostasia primaria [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] Stefanini L, Paul DS, Robledo RF, Chan ER, Getz TM, Campbell RA, et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J Clin Invest 2015;125:1419–32. Jackson SP, Nesbitt WS, Westein E. Dynamics of platelet thrombus formation. J Thromb Haemost 2009;7:17–20. Gresele P, Harrison P, Bury L, Falcinelli E, Gachet C, et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost 2014;12:1562–9. Fressinaud E. Diagnostic clinique, biologique et moléculaire de la maladie de Willebrand. Hematologie 2014;20:36–49. Harrison P, Mackie I, Mumford A, Briggs C, Liesner R, Winter M, et al. Guidelines for the laboratory investigation of heritable disorders of platelet function. Br J Haematol 2011;155:30–44. Gresele P. Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis. Diagnosis of inherited platelet function disorders: guidance from the SSC of the ISTH. J Thromb Haemost 2015;13:314–22. Provan D, Stasi R, Newland AC, Blanchette VS, Bolton Maggs P, Bussel JB, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86. Canault M, Ghalloussi D, Grosdidier C, Guinier M, Perret C, Chelghoum N, et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med 2014;211:1349–62. Lowe GC, Lordkipanidzé M, Watson SP, The UK GAPP study group. Utility of the ISTH Bleeding Assessment Tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost 2013;11:1663–8. Mumford AD, Frelinger 3rd AL, Gachet C, Gresele P, Noris P, Harrison P, et al. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb Haemost 2015;114:14–25. Eto K, Kunishima S. Linkage between the mechanisms of thrombocytopenia and thrombopoiesis. Blood 2016;127:1234–41. Bosticardo M, Marangoni F, Aiuti A, Villa A, Grazia Roncarolo M. Recent advances in understanding the pathophysiology of WiskottAldrich syndrome. Blood 2009;113:6288–95. Nurden AT, Nurden P. Inherited disorders of platelet function: selected updates. J Thromb Haemost 2015;13(Suppl. 1):S2–9. Mattina T, Perrotta CS, Grossfeld P. Jacobsen syndrome. Orphanet J Rare Dis 2009;4:9. Pallotta R, Evangelista V, Margaglione M, Bucci I, Saponari A. Macrothrombocytopenia in velocardiofacial syndrome. J Thromb Haemost 2005;3:601–3. Ballmaier M, Germeshausen M. Advances in the understanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol 2009;146:3–16. [20] Antony-Debré I, Manchev VT, Balayn N, Bluteau D, Tomowiak C, Legrand C, et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood 2015;125:930–40. [21] Noris P, Favier R, Alessi MC, Geddis AE, Kunishima S, Heller PG, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood 2013;122:1987–9. [22] Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 2015;47:535–8. [23] Balduini CL, Savoia A, Seri M. Inherited thrombocytopenias frequently diagnosed in adults. J Thromb Haemost 2013;11:1006–19. [24] Fletcher SJ, Johnson B, Lowe GC, Bem D, Drake S, Lordkipanidze M, et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clin Invest 2015;125: 3600–5. [25] Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14. [26] Levade M, David E, Garcia C, Laurent PA, Cadot S, Michallet AS, et al. Ibrutinib treatment affects collagen and von Willebrand factordependent platelet functions. Blood 2014;124:3991–5. [27] Bianchi E, Norfo R, Pennucci V, Zini R, Manfredini R. Genomic landscape of megakaryopoiesis and platelet function defects. Blood 2016;127:1249–59. [28] Turro E, Greene D, Wijgaerts A, Thys C, Lentaigne C, Bariana TK, et al. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci Transl Med 2016;8, 328ra30. [29] Malfait F, De Paepe A. Bleeding in the heritable connective tissue disorders: mechanisms, diagnosis and treatment. Blood Rev 2009;23:191–7. [30] Lentaigne C, Freson K, Laffan MA, Turro E, Ouwehand WH. Inherited platelet disorders: toward DNA-based diagnosis. Blood 2016;127:2814–23. Si desea saber más Hézard N, Simona G, Droulleb A, Nguyen P. La cytométrie de flux dans un laboratoire d’hémostase. Rev Fr Lab 2007;393:63–71. Favier R, Bardet V, Khorsi S, Adam M. Le diagnostic des thrombopénies constitutionnelles. Rev Fr Lab 2006;378:35–42. Payrastre B, Alessi MC, Sie P. Physiopathologie des thrombopathies constitutionnelles. Hematologie 2014;20:20–35. Protocole national de diagnostic et de soins sur le purpura thrombopénique immunologique de l’enfant et de l’adulte. Octobre 2009. A. Rauch, Praticien hospitalier (antoine.rauch@chru-lille.fr). Institut d’hématologie-transfusion, Centre de biologie pathologie génétique, CHU de Lille, 59037 Lille cedex, France. Inserm U1011, Récepteurs nucléaires, maladies cardiovasculaires et diabète, Laboratoire de recherche J & K, Faculté de médecine, Pôle recherche, boulevard du Professeur-J.-Leclercq, 59045 Lille cedex, France. C. Paris, Assistante hospitalo-Universitaire. Institut d’hématologie-transfusion, Centre de biologie pathologie génétique, CHU de Lille, 59037 Lille cedex, France. Cualquier referencia a este artículo debe incluir la mención del artículo: Rauch A, Paris C. Hemostasia primaria. EMC - Tratado de medicina 2017;22(1):1-12 [Artículo E – 1-1272]. Disponibles en www.em-consulte.com/es Algoritmos 12 Ilustraciones complementarias Videos/ Animaciones Aspectos legales Información al paciente Informaciones complementarias Autoevaluación Caso clinico EMC - Tratado de medicina